Bağışıklık sisteminin merkezi hücresi lenfosittir. Lenfositler uygun immün (bağışık) yanı ta aracılık ederler. Özgün patojenlere karşı yanıt oluşturarak bağışıklık sistemine özgünlük kazandırır ve reinfeksiyona karşı uzun süreli bağışıklık geliştirirler. Lenfositler kemik iliğinde yerleşmiş ve kanın tüm hücresel elemanlarına kaynak oluşturan pluripotent hematopoietik kök hücrelerinden köken alır. Lenfositlerin iki ana işlevsel sınıfı vardır: (I) B lenfositleri veya B hücreleri ve (2) T lenfositler veya T hücreleri, gelişme yerleri, antijenik reseptörleri ve işlevlerine göre ayırt edilirler. Lenfositlerin başlıca bozuklukları şunlardır: (1) Lenfoma veya lösemi gelişmesiyle sonuçlanan lenfositlerin alt yapılarının neoplastik transformasyonu, (2) İmmün yetmezlik sendromları ile sonuçlanan lenfosit gelişmesi ve işlevlerinde konjenital ve edinsel kusurlar, (3) Lenfadenopati, lenfositoz veya Ienfositopeniye yol açan antijenik uyarı veya enfeksiyona fizyolojik yanıt.
B hücreleri hücre yüzey immünglobulinleri (veya antikorları) varlığı ile karakterizedir. Asıl işlevleri antijene yönelik özgül antikor üreterek antijenlere karşı hümoral immün yanıt meydana getirmektir. B hücreleri kemik iliğinde yüksek derecede koordine edilen ağır ve hafif zincir immünglobulin genlerinin ardışık olarak yeniden düzenlenmesi ve B hücrelerine özgü yüzey proteinlerinin üretilmesini kapsayan bir seri basamaktan geçtikten sonra gelişirler. İmmünglobulin genlerinin tekrar düzenlenmesi (rearrangement) büyük miktarda B hücre çeşitliliği oluşturur. Her bir B hücresi çeşidi kendine özgü antijenik spesifite taşır. B hücreleri kemik iliğinden tüm vücut içersine dağılmış haldeki lenfoid dokuya göç ederler. Hücre yüzey immünglobulinleri ve antijenleri varlığı ile hemen ayırt edilir. Bunlar B hücrelerine özgül olup CD 19, CD20 ve CD21’i içermektedir. Hücre yüzey immünglobulinlerine bağlanan antijenlere yanıt olarak matür B hücreleri çoğalmak üzere aktive olup, son aşama plazma hücrelerine (end-stage plasma cells) farklılaşma geçirirler. Büyük miktarlarda çözünebilen antikorlar üretirler. B hücrelerinin neoplastik bozuklukları farklı gelişme safhalarındaki B hücrelerinden kaynaklanır. Böylece B hücre lenfomaları morfolojik mekanizmaları ve hücre yüzey ekspresyonları veya immünfenotipleri bakımından çok fazla çeşitlilik gösterir.
T hücreleri, klasik olarak hücresel bağışık yanıt olarak adlandırılan bir dizi işlevleri gerçekleştirir. T hücre öncü ileri kemik iliğinden timusa göç ederek olgun T hücre altyapılarına farklılaşırlar. Vücudun kendi peptidlerine yanıt veren otoreaktif T hücrelerini yok etme yeteneği kazanır. Timusta T hücre öncülleri T hücre reseptör (TCR: T-cell receptor) genlerinin tekrar düzenlenmesi ve ekspresyonu ile CD3, CD4 ve CD8 gibi sadece T hücrelere özgül hücre yüzey proteinlerin kazanılmasını içeren düzenlenmiş bir farklılaşma süreci geçirirler. T hücreleri timusta olgunlaşır iken CD4 veya CD8 proteinlerinden birini kayıp eder, ve böylece olgun T hücreleri iki grup haline gelir: CD4+ ve CD8+ hücreler. Timusta T hücre ayrımlaşması ve olgunlaşmasından sonra, olgun CD4 ve CD8 T hücreleri timustan çıkıp lenf düğümleri, dalak ve periferik bağışıklık sistemine ait alanlara göç ederler. Olgun T hücreleri periferik kan lenfositlerinin yaklaşık olarak %80’ini, lenf düğümündeki hücrelerin %40’ını ve dalaktaki lenfoid hücrelerin %20’ini oluştururlar. Olgun CD4 ve CD8 T hücre alt birimleri açıkça belirlenmiş, birbirinden ayırt edilmiş bağışıklık işlevlerine aracılık ederler. CD8+ hücreler virüsle enfekte olmuş hücreleri veya yabancı hücreleri öldürür ve bağışıklık işlevlerini baskılar; CD8 hücreler sitotoksik T hücreleri olarak adlandırılır. CD4+ hücreler, sitokinler üreten ve doğrudan hücrelere temas oluşturarak malıofajlar ve B hücreleri gibi bağışık yanıt hücrelerini aktive ederler; bu yüzden CD4 hücreleriyardımcı T hücreleri olarak adlandırılır. B hücrelere benzer biçimde, T hücreleri kendilerine özgü TCR molekülleri oluşturur. B hücrelerin aksine, T hücreleri sadece hücre içerisinde işlenmiş ve özelleşmiş hücre yüzey antijeni sunan proteinlere bağlanan peptidlere yanıt verirler. Bu petidIere major histokompatibilite kompleksi: MHC denir. CD4 ve CD8 hücreleri peptid-MHC kompleksIerine yanıtlarında MHC-sınıfıyla kısıtlıdırlar. CD4 hücreleri antijenik peptid parçalannı sadece MHC sınıf IImolekülleri tarafından sunulmuşlarsa tanıyabilirler. CD8 hücreleri de antijenik parçalan sadece MHC sınıf i molekülleri tarafından sunulmuşlarsa tanıyabilirler. MHC sınıf 1 ve 2 molekülleriyle kompleks oluşturan antijenik peptidler farklı kaynaklardan köken alırlar. MHC sınıf 1 moleküller genellikle hücre içi veya endojen antijenleri üretir, sitozolde işlenir ve endoplasmik retikulumdan geçerek dağılırlar. MHC sınıf 2 molekülleri ise genellikle endositoz ile hücre içine alınıp hücre içi veziküllerde işlenen hücre dışı kaynaklı antijenleri üretir. Özgül peptid-MHC kompleksi ile T hücre reseptörünün bağlanması aktivasyon sinyallerini tetikleyerek gen ürünlerinin ekspresyonuna yol açarlar. Bunların sonucunda CD4+ hücrelerin yardımcı veya CD8+ hücrelerin sitotoksik işlevlerinin çok geniş çeşitliliği ortaya çıkar.

Lenfositler, antijen-lenfosit karşılıklı etkileşim ve lenfosit aktivasyon yeri olan periferik lenfoid dokuda yerleşirler. Periferik lenfoid doku; lenf düğümleri, dalak ve mukozal lenfoid dokudan oluşmaktadır. Lenfositler vasküler ve lenfatik sistemler boyunca sürekli dolaşımdadır Lenf düğümleri yüksek derecede organize lenfoid dokulardır. Buralar lenfatik drenaj sisteminin birleşme yerleri olup drenajı yapılmakta olan lenfatikten lenf düğümüne antijen taşınır ve lenf düğümlerinde bu antijenler tutulurlar. Bir lenf düğümünü dışta korteks ve içte medullan oluşmaktadır. Korteks baskın bir şekilde B hücrelerinden oluşan lenfoid foliküllere organize olmuştur. Bazı foliküller merkezi alanlar veya germinal merkezleri içerir ve burada özgül bir antijenle karşılaşan aktive B hücreleri çoğalma faaliyeti içersindedir. Dış taraftan mantle zone (manto bölgesi) ile çevrilidir. T hücreleri folikülleri saran parakortikal bölgelerde daha yaygın olarak dağılmışlardır. Dalak lenfatiklerden ziyade kan damarlarındaki antijenleri yakalar. eritrositlerin yaşlandıklarında atılma alanını oluşturur. Dalaktaki lenfositler beyaz pulpa olarak adlandırılan bölgelerde yerleşmişlerdir. Beyaz pulpa organa gelen arteriyolleri çevreler. Lenf düğümlerinde olduğu gibi, B ve T hücreleri periarteriyoler lenfoid kılıf içinde dağılmış halde bulunurlar. Bu kılıf T hücrelerden ve onu kenarlardan destekleyen foliküller ise B hücrelerden oluşmaktadır. Mukoza ilişkili lenf dokuları (mucosa associated lymphoid tissues-MALTs) epitelyal yüzeylerden antijen toplar. Mukozal bölgelerdeki daha yaygın şekilde organize olmuş lenfosit kümeleri de dahil olmak üzere, gastrointestinal sistem ilişkili lenfoid dokuyu [gut-associated lymphoid tissue] (bademcikler, adenoidler, appendiks ve ince barsaktaki peyer plakları) da içerir. Lenfositler periferik kan dolaşımındadır ve erişkinlerde periferik kan lökositlerinin %20-40’ını oluşturur (bu oran yenidoğanlarda ve çocuklarda daha yüksektir). Periferik kan lökositlerinin %80-90’ı T hücreleridir ve geri kalanı ise çoğunlukla B hücreleridir. Periferik kan dolaşımındaki lenfositlerinin çoğu olgundur, geri kalan lenfositler morfolojik olarak küçük olup sitoplazmaları çok dardır ve nükleolusları fark edilmeyecek şekildedir. Periferik kan lenfoid hücrelerinin küçük bir oranı natural killer (NK) hücreler olarak adlandırılan üçüncü bir kategoriyi temsil eder. NK hücreleri B veya T hücrelerin karakteristik hücre yüzey moleküllerini taşımazlar. Bunların immünglobulin veya T hücre reseptör (TCR) genleri yeniden düzenleme sürecini gerçekleştirınezler. Morfolojik olarak bu hücreler büyük azurofilik granüller içeren bol sitoplazmalı, büyük granüllü hücreler olarak adlandırılır. İşlevsel olarak, doğal bağışıklık sisteminin bir parçasıdır. Birincil antijenik temas gerektirmeksizin birçok patojen grubuna özgül olmayan yanıt verebilmektedir.
Lenfositlerin malign dönüşümleri çok çeşitli lenfoid kökenli neoplazmalara yol açar. T veya B hücrelerden kaynaklanan tümörler ve lenfosit gelişiminin farklı safhalarını temsil eden tümörleri içerirler. Lenfoid malignensileri genellikle lenfoid dokuları içerir, fakat herhangi bir yerden köken alabilirler veya yayılabilirler. Lenfoid malignensilerin ana grupları non-Hodgkin lenfomalar veya hodgkin dışı lenfomalar (non-Hodgkin’s lymphomas, NHL), Hodgkin hastalığı (Hodgkin’s disease, HD), lenfoid lösemiler ve plazma hücre diskrazileridir. Erişkinlerde lenfoid bir malignansinin en yaygın klinik görünümü ağrısız lenf nodu büyümesi veya lenfadenopatidir. Lenfadenopatinin lenfoid malignansilerin yanı sıra birçok nedeni vardır. Bu yüzden iyi bir hastalık öyküsü alınması ve dikkatli bir fizik muayene yapılması lenf düğümü biyopsi işleminden önce yerine getirilmesi gereken önemli işlemlerdir. Lenfadenopatinin araştırması büyümüş lenf düğümünün yerleşimine (bölgesel veya yaygın) ve klinik semptomların varlığına göre düzenlenebilir. Servikal lenfadenopatiler en sık üst solunum yolu enfeksiyonları tarafından meydana gelir. Bunun nedenleri arasında bakteriyel faranjitin yanı sıra, enfeksiyöz mononükleoz ve diğer viral sendromlar da yer almaktadır. Unilateral aksiller, inguinal veya femoral adenopati kedi tırmığı hastalığı dahil ekstremiteyi içeren deri enfeksiyonlarında ortaya çıkabilir. Yaygın lenfadenopati sistemik enfeksiyonlardan kaynaklanabilir, örneğin insan immün yetmezlik virüsü (human immunodeficiency virus, HIV) veya sitomegalovirüs (cytomegalovirus, CMV) enfeksiyonu, ilaç reaksiyonları, otoimmünhastalıklar, sistemik lenfadenopati sendromlanndan biri veya lenfoma olabilir. İyi bir değerlendirmeden sonra inatçı lenfadenopatinin sebebi bulunamamışsa, lenf düğümü biyopsisi yapılmalıdır. Büyümüş bir suprakJavikular lenf düğümü yüksek olasılıkla malignensi düşündürür ve her zaman biyopsi örneklemi alınması gerekir. Lenfomanın kesin tanısı lenf düğümünün eksizyonel biyopsisini veya etkilenen lenf dokusundan genişçe bir biyopsi yapılmasını gerektirir. İnce iğne aspirasyonu veya iğne biyopsi si malign lenfomalaıın tanısında nadiren yeterli olmaktadır. Alınan örneğin patolojik incelemesi rutin histolojik incelemeyi ve immünfenotiplendirmeyi içerir. İmmünfenotiplendirme malign lenfositin üzerinde görülen immünolojik hücre yüzey antijenlerinin monoklonal antikorlar yardımıyla tanımlanmasını içerir. İmmünfenotiplendirme hücrenin hangi çeşit olduğunu (B hücresi, T hücresi, NK hücresi veya ekstralenfoid hücre) ve hücre yüzeyantijenlerinin sıralanışınm tanılanmasına izin verir. B hücreli NHL’ larda, immünfenotiplendirme yöntemi yüzey immünglobulinleri kappa veya lambda hafif zincirlerinden oluşup oluşmadığını belirleyerek, bu durumun kökeninde monoklonal bir süreç olup olmadığını açıklar. İmmünfenotiplendirme lenfomalann tanısında ve sımflandırılmasında mutlaka bulunması gereken bir işIemdir. İmmünfenotiplendirıne, doku örneklerinde immünhistokimyasal ve f10w sitometrik analizler yapılarak gerçekleştirilebilir.
Bazı olgularda, T hücre reseptör gen yeniden ayarlanması veya immünglobulinleri gösteren sitogenetik analiz ve moleküler çalışmalar, lenfomanın patolojik alt tiplendirmesini yapmak veya monoklonal bir süreci (örn., malign) ortaya koymak için gerekebilir. Eğer yapılan bir lenf düğümü biyopsisi tanılanamamışsa ve açıklanamayan lenf düğümü büyümesi devam ediyorsa, biyopsi yinelenmelidir. Hodgkin dışı lenfomalar (Non Hodgkin Lenfomalar, NHL, HO ) NHL, Histolojik görünümlerine, hücre kökenlerine, prognozlarına ve tedaviye yanıtlarına göre değişen bir grup heterojen lenfoid malign neoplazm türüdür. NHL’ların heterojenliğini göz önünde tutarak, farklı klinik yansımalara paralel özgül patolojik alt tipleri tanımlamak için sınıflandırma sistemleri geliştirilmjştir. Histopatolojik ve biyolojik davramşlar arasındaki korelasyonlar ortaya kondukça, son elli yıldan fazla bir sürede bu sistemler yavaş yavaş gelişebilmiştir. Yakın zamana kadar Kuzey Amerika’da en yaygın kullanılan sınıflandırma sistemi (Uluslararası) Çalışma Formülasyonuydu (ÇF, International Worbng Formulation). ÇF lenf düğümünün yapısına (foliküllerin mi yoksa yaygın infiltratın mı daha çok olduğuna) ve maljgn lenfositlerin morfolojik özelliklerine (bölünmüş veya bölünmemiş nükleuslu, küçük veya büyük hücreli) göre NHL’ları sınıflandırdı ve doğal öyküleri ve klinik davranışlarına göre düşük, orta ve yüksek dereceli olarak patolojik alt tiplerine ayrımnı yapmıştı. Genel olarak düşük dereceli histolojik yapılar, yavaş bir seyir ve görece uzun bir sağ kalım, fakat tedavi edilemez bir klinik durum oluştururken, orta ve yüksek dereceli histolojik yapılar, biyolojik olarak agresif (yani tedavi edilmezse kısa yaşam seyri olan), fakat uygun tedaviyle potansiyel olarak sağaltılabilir bir seyir gösteriyordu. Lenfomaların immünfenotiplendirilmesi ve moleküler olarak karakterizasyonu sonucunda görülmüştür. ÇF özgül patolojik ve klinik oluşumları tam olarak tanımlayamamıştır. 1994’de tarif edilen, Revised European-American Lymphoma (REAL- Gözden geçirilmiş Avrupa-Amerikan Lenfoma) sınıflandırma sistemi sadece, ÇF’da tanımlanan, hjstolojik özellikleri değil, ayrıca immünfenotiplendirme, sitogenetik, ve epidemiyolojik ve etiyolojik özellikleri de tanımlamaya dahil etmiştir. Böylece, REAL sınıflandırması ÇF’ da kolaylıkla sınıflandırılamamış olan çeşitli NHL alt tiplerini tanımlamıştır.
Bu alt tipler, mantle celllenfomaları içerirler; MALT lenfomaları (Mucosa Associated Lymphoid Tumor, mukoza ile bağlantılı lenfoid tümör) ve monositoid B hücreli lenfomaları, ki bu ibsi de lenf düğümlerinin marjinal bölgelerinden köken alırlar, ve insan T hücreli lösemi virüsü tip i aracılı lösemi (human T cell leukemia virus type 1- associated leukemia) dahil çeşitli T hücreli lenfomaları, kütanöz T hücreli lenfomaları (mikozis fungoides veya sezary sendromu) ve biyolojik olarak agresif periferik T hücreli lenfomalardır. REAL sınıflaması 2001 'de WHO-Dünya Sağlık Örgütü tarafından güncelleştirilnuş ve REAL WHO sınıflaması tüm önceki sınıflandırma sistemlerinin yerini almıştır. ABO’de en sık görülen NHL; foliküler lenfomalar, küçük lenfositik lenfoma veya lösemi (kronik lenfositik lösemi-KLL olarak ta bilinir), mantle reel 1 lenfomalar ve yaygın büyük B hücreli lenfomalarıdır. NHL’ların çoğunun nedeni bilinmemektedir. NHL’lı hastaların çoğunluğunda belirgin bir genetik yatkınlık veya epidemjyolojik
veya çevresel etmen tanımlanamamıştır. NHL alt tiplerinin çoğu patogonomik kromozamal translokasyonlar taşırlar b sıklıkla bir immünglobulin loküs (veya T-hücre kökenli NHL’larda T hücre reseptör loküs) ve bir onkogen veya büyüme düzenleme geni (growth regulatory gene) içerir. Bu aberan kromozamun yeniden düzenlenmesinin nedeni bilinmemektedir. Konjenital immün yetmezlik sendromu veya otoimmün bozuklukları olan hastalar NHL geliştirme konusunda yüksek risk taşırlar. Seyrek görülen HL türlerinden bazılannın meydana gelmesinde onkojenik insan virüsleri rol oynarlar. Ebstein Ban Virus (EBV) biyolojik olarak agresif birkaç NHL ile bağlantılıdır: Edinsel bağışıklık yetmezlik sendromu-AIOS- bağlantılı yaygın agresif lenfomalar, organ nakli sonrasında immün baskılanmış hastalarda gelişen immün proliferatifbozukluklar ve Afrika’da endemik olan Burkitt lenfonu HTLV -1, T hücre löseminin agresif bir
formuyla veya Japonya ve Karayip adalannda endemik olarak görülen lenfomayla sebep-sonuç bağıntısı vardır. Kaposi sarkomadab herpes virüsün, serozal kavitelerde gelişen yaygın bir agresif NHL’nın bir çeşidi ile bağlantısı olduğu gösterilmiştir ve HIV ile enfekte hastalarda bu virüse yaygın bir şekilde rastlanınaktadır. Helicobacter pylori enfeksiyonun gastrik MALT lenfomaları ile sebep-sonuç ilişbsi vardır ve antibiyotiklerle bu enfeksiyonun eradikasyonu sıklıkla lenfomanın gerilemesiyle sonuçlanmaktadır.
Bir önceb bölümde anlatıldığı üzere, NHL’lı hastaların çoğunluğu bir veya daha fazla periferik lenf düğümü bölgesini ilgilendiren ağrısız lenfadenopati ile kendilerini gösterirler. Ek olarak, NHL düğüm dışı alanları tutabilir, böylece tutulan bölgelere ait bir takım yakınmaları gösterebilirler. Gerçekte NHL herhangi bir alanı potansiyel olarak tutabilmekle birlikte, ekstranodal hastalığın en çok tuttuğu alanlar gastrointestinal sistem, kemik iliği, karaciğer ve Waldeyer halkasıdır. Genelde, NHL’nın agresif alt grupları (yaygın büyük hücreli, lenfoblastik, ve Burkitt ) ekstranodal alanları agresif olmayanlardan daha fazla tutar. Leptomeningeal yayılım dahil, merkezi sinir sistemi tutulumu agresif olmayanlarda ender görülürken, agresif olanlarda beklenen bir durumdur. En agresif NHL’lar (Burkitt ve lenfoblastik) leptomeninkslere özellikle yayılım gösterme eğilimindedir. Ateş, kilo kaybı, gece terlemeleri gibi yapısal yakınmalar tanı konduğunda NHL’lı hastaların %20’sinde görülür. NHL’ların agresif alt gruplarında bu yakınmalara daha sık karşılaşılır.NHL tanısı tutulan lenf düğümünün veya düğüm dışı alandan biyopsi yapılmasını gerekli kılar. Küçük lenfositik lenfoma veya honik lenfositik lenfomada olduğu gibi, kemik iliği ve periferik kan tutulumu olan hastalarda periferik kan lenfositlerinin f]ow sitometri ile immünfenotip tanısının gerçekleştirilmesi mümkündür. Lenfoma tanısı bir kez konduktan sonra, hastaların tam bir evrelendirilmesi gereklidir. Evrelendirme tutulumun yaygınlığını belirler, prognozu hakkında bilgi verir ve tedavi seçimini etkileyebilir.
Modifiye Ann Arbor evrelemesi hem NHL’nın, hem de Hodgkin hastalığının evrelemesinde kullanılmaktadır. Standart evrelerne değerlendirmesi, yapısal yakınmaların varlığını sorgulayan (ateş, gece terlemesi veya kilo kaybı, B semptomları olarak adlandırılır) lenfomayı düşündürecek yakınmalan ortaya çıkaracak iyi bir öykü; büyümüş lenf düğümlerinin ebatlarını ve dağılımlarını gösteren tam bir fizik muayene; laktat dehidrogenaz (LDH) dahil kan testlerini; göğüs, batın ve pelvis bilgisayarlı tomografisini; kemik iliği aspirasyonu ve biyopsisini içerir. Galyum veya pozitron emisyon tomografi taramalarıagresif HL’ların evrelendirilmesinde sıklıkla araştırmaya dahil edilmekle birlikte, galyum tutan (galyum avid) veya metabolik olarak aktif (sıklıkla agresif alt gruplar; örn., yaygın büyük hücreli lenfoma, lenfoblastik lenfoma veya Burkitt lenfoma) lenfomaların tedaviye yanıtlarının değerlendirilmesinde de yardımcı olabilmektedir. Sitolojik analiz için lomber ponksiyon sadece Burkitt lenfoma ve lenfoblastik lenfomalı tüm hastalar ve kemik iliği, testis veya merkezi sinir sistemine doğrudan bitişik yapılara (örn., paranasal sinüsler, calvarium) tutulum gösteren yaygın büyük hücreli lenfomaları içeren leptomeningeal yayılım riski olan hastalarda yapılmalıdır. Özel durumlarda yardımcı testler yapılabilir. Örneğin, erişkin T-hücreli lösemi/lenfoma veya AIDS-ilişkili lenfoma olduğu düşünülenlerde, sırasıyla HTLV-I veya HIV testleri yapılmalıdır. Gastrointestinal sistem yakınmaları olanlar veya GİS tutulum riski olan (Waldeyer halkasını tutan) tüm hastalarda GİS testleri ve endoskopik değerlendirmesi gerekebilir. Serum protein elektroforezi ve 2-mikroglobulin ve immünglobulinlerin niceliksel değerlendirilmesi plazma hücre diskrazileri olduğu düşünülen hastalarda yapılmalıdır. NHL’lı hastalarda sadece evrelendirme amacıyla laparatomi hiçbir zaman yapılmamalıdır, çünkü seçilecek tedavi kararını nadiren etkilemektedir. NHL için bir grup prognoz belirleyici değişkenler tanımlanmıştır. Genel olarak, NHL’ların alt gruplarında kötü sağkalım öngördürücüleri tanı konduğunda ileri evre oluşu (evre III veya IV), birden çok ekstranodal alan tutulumu, yüksek LDH düzeyleri, B yakınmalarının varlığı ve kötü efor kapasitesidir.
Nonagresif NHL’lar Basit düşük dereceli veya yavaş seyirli histolojik durumlar foliküler lenfomalar (küçük bölünmüş hücreli ve karma hücre tipleri) ve küçük lenfositik lenfomadır (bu sonuncusu KLL ile aynıdır ve daha sonra tartışılacaktır). Tüm NHL’ların sırasıyla yaklaşık olarak %30 ve %5’ine karşılık gelirler. Düşük dereceli foliküler lenfomalar, yüzey immünglobulinleri (kappa veya lambda kısıtlı) ve olgun B-hücre belirleyicileri (CD19, CD20, CD21) pozitif, CD5 için negatif immünfenotiplendirmeleri olan olgun klonal B-hücre neoplazmlarıdırlar. Foliküler lenfomalar sitogenetik olarak t(l4;18) translokasyonuyla karakterizedirler ki bu translokasyon foliküler lenfomalarda BCL2; BCL2 antiapoptotik genlerle immünglobulin ağır zincirlerinin yan yana bitişik sıralandığı bir örnek şekilde eksprese edilir. Foliküler lenfomalar düşük dereceli olmalarına karşın, yavaş seyirli neoplazmlar olup, uzun hastalık gelişim öyküleri vardır (medyan sağ kalım LO yıla ulaşır), hastaların çoğu (%80-90’ı) ileri evredirler (evre III veya IV), sıklıkla standart tedavi usulleriyle sağaltılamazlar. Kısa sağ kalımla ilişkili etmenler ileri yaş, ileri evre, anemi, çoklu lenf düğümü alanlarının tutulumu (dörtten az) ve yüksek LDH düzeyleridir. Bu etmenlerden üçü veya daha fazlasını taşıyan hastaların medyan sağ kalımları 5 yıl olup, bu süreç risk faktörlerinden birini taşıyan veya hiç olmayan hastaların kabaca yarı süresidir. Foliküler NHL’lı hastaların çoğu daha agresif lenfomaya dönüşüm gösterir; patolojik olarak yaygın büyük hücre infiltrasyonu vardır, klinik olarak lenf düğümleri veya diğer tümör kitleleri hızla büyüme gösterir, LDH düzeyleri yükselir ve hastalığa bağlı yakınmalar başlar.
Foliküler lenfomaların tedavisini bulunduğu evre belirler. Klinik evreleme sonrası erken evreli (I veya büyük kitleli olmayan II) oldukları kabul edilen az sayıdaki hastada, uygun tedavi radyoterapidir. Subtotal veya total lenfoid radyoterapi ile erken evre hastaların yarısından fazlası sürekli bir remisyon gösterir ve kür kabul edilebilirler. İleri evre hastalarda tedavi yaklaşımı biraz tartışmalıdır. İleri evre yavaş seyirli NHL bir grup tedavi seçeneklerine yanıt vermekle birlikte, tedaviye direnç göstermesi ve uzun doğal seyirli oluşu hasta semptom geliştirinceye kadar tedavinin ertelenmesini gerektirmiştir. Bu strateji bekle ve izle yaklaşımı olarak kabul edilir.

Tedavi endikasyonları büyümüş lenf düğümlerinden kaynaklanan kozmetik veya mekanik sorunlar, hastalığı düşündürecek yapısal semptomlar (ateş, kilo kaybı, gece terlemeleri gibi), kemik iliği etkilenimi gibi bulguları içerir. İleri evre hastalıkta gerekli olduğunda uygun tedavi, sistemik kemoterapidir. Foliküler lenfomalar tekli ve çoklu ilaç tedavi programlarına yanıt verir. Alkilleyici ajanlardan tekli (siklofosfarnid veya klorambusil), bir alkilleyici ajan içeren çoklu ilaç rejimieri (örn., siklofosfamid, vinkristin, ve prednizolon [CVPJ), veya fludarabin esaslı rejimler (yani, fludarabin ve mitoksantron, fluda-rabin ve siklofosfamid) bu hastalıkta etkin başlangıç tedavi rejimIeridir. Kemoterapiye kimerik (fare-insan) anti-CD 20 monoklonal antikorı Rituksimab ilavesi yaygınkabul görmüş bir uygulamadır, yanıt hızında ve remisyon süresinde iyileşmeler sağlamıştır. Hastaların çoğunluğu tedaviye yanıt vermektedir, en azından üçte birinde 1-3 yıl süren tam klinik remisyon başarısı sağlanmaktadır. Maksimum yanıta ulaşıldığında kümülatif toksisiteyi en aza indirgemek için tedavi durdurulmalıdır. Hasta relaps gösterirse, ardıl remisyonlar başarılabilir, fakat ilk remisyona göre genellikle daha kısa sürelidir. Relaps gösteren hastalar için tedavi seçenekleri sıklıkla ilk tedaviden farklı bir ilaçla veya farklı bir kombinasyon içeren kemoterapiyle tekrar tedavi etmektir. Relapstaki hastalar tek ajan olarak Rituksimab ile tedavi edilebilirler. Relaps gösteren foliküler lenfomalı hastalarda Rituksimab oldukça etkin, toksik olmayan bir ajan olup, hastaların yarısından fazlasında sıklıkla devam eden yanıtlar oluşturur. Rituksimaba yanıt veren hastalar yineleyen relapslarda sıklıkla başarılı olarak yine rituksimabla tekrar tedavi edilebilirler. İkinci veya üçüncü sıra kemoterapiyle edinilen deneyimlerin aksine, bu hastalar rituksimabla edindikleri ilk remisyondan daha uzun süren remisyon süreleri göstermektedirler. Radyoaktif işaretli anti-CD20 antikorları olan ibritumomab tiuksetan (yytrium-işaretli) ve iyot-Bl tositumomab relaps gösteren veya refrakter folliküler lenfomalı hastalarda kullanılmakta ve yüksek yanıt oranları elde edilmiştir. Klinik veya patolojik olarak daha yüksek dereceli lenfomaya dönüşüm gösteren hastalarda, yaygın agresif histoloji gösteren hastalık için uygun olan tedavi önerilmelidir (daha sonra tartışılacaktır). Foliküler NHL’larda otolog veya allogen kök hücre transplantasyonu ile birlikte yüksek doz kemoterapinin rolü halen tam açıklığa kavuşmamıştır ve deneysel bir yaklaşım olarak değerlendirilmelidir.
Allogen transplantasyon uygulanan hastaların uzun dönem izlemi bu tedavi yönteminin bazı hastalarda kül’ sağladığını düşündürmektedir. Ancak, allogen transplantasyonla ilişkili komplikasyon, refrakter yavaş seyirli lenfomalarda bu tedavinin yaygın kullanımını kısıtlamıştır. Foliküler NHL’lara ek olarak, MALT lenfomalar ve bununla yakından ilişkili marginal bölge lenfomaları da düşük dereceli agresif olmayan alt tipler olarak değerlendiriImektedir. Mükemmel prognoz, bölgeseloluşları ve uzun doğal seyir göstermeleri nedeniyle MALT lenfomalarda sistemik kemoterapiden kaçınılmakta ve bölgesel tedavi yöntemleri ile (radyoterapi veya ameliyat) genellikle koruyucu yaklaşım uygulanmaktadır. Gastrik MALT lenfomaların H. Pylori enfeksiyonu ile yüksek oranda ilişkili olması önemli bir bilgi olup, bu enfeksiyonun tedavi edilmesi sonucunda remisyonlar sıklıkla sağlanabilmektedir. Bu yüzden, erken gastrik MALT lenfomada antibiyotik uygulaması ilk sıra tedavisidir.
Agresif NHL’lar büyük lenfositlerin yaygın infiltrasyonu sonucunda lenf düğümü yapısının silinmesi ile karakterizedir. Yaygın büyük B-hücreli lenfoma (bütün NHL’ların üçte birini oluştururlar), anaplastik geniş hücreli lenfoma ve periferik T-hücre lenfomayı içerir. Yaygın agresif geniş hücreli lenfomaların çoğu B hücre kökenlidir (yaygın geniş B-hücreli lenfoma); T-hücreli yaygın agresif lenfomalar veya periferik T-hücreli lenfomalar benzer şekilde değerlendirilirler, fakat B-hücreli benzerlerinin aksine genelde kötü prognoz gösterirler. Burkitt lenfoma ve lenfoblastik lenfoma en agresif lenfomalar arasındadır ve bu bölümde daha sonra ayrıca tartışılacaktır (yüksek dereceli NHL’lara bakınız). Yaygın agresif NHL’lar klinik olarak agresif davranış gösterir ve eğer tedavi edilmezlerse medyan sağ kalım 1-2 yıldan azdır. Foliküler NHL’larla karşılaştırıldığında, yaygın agresif histolojisi olan hastaların büyük bir bölümü erken evre hastalığı (%30-50) veya ekstranodal alan tutulum ile (%50) kendini gösterir. Yaygın agresif histolojileri olan hastaların kül’ şansı ve sonuçları hastalık ortaya çıktığında görülen olumsuz prognostik özelliklerine doğrudan bağlıdır: 60 yaş üstü, ileri evre (III veya IV), yüksek LDH düzeyleri, kötü efor kapasitesi ve iki veya daha fazla ekstranodal alanda hastalık oluşu. Kül’ veya uzun süreli hastalıktan muaf sağ kalım şansı, bir veya birkaç kötü prognoz faktörleri olan hastaların %75’inden fazlasında görülürken, dört veya daha fazla kötü prognoz faktörü olanların %30’undan azında görülmektedir. Düşük dereceli foliküler NHL’lı hastaların aksine, yaygın agresif histolojileri olan hastaların hepsine tedavi hemen verilmelidir. Çünkü bu lenfomalarda potansiyel olarak kül’ sağlanabilir. Yaygın agresifNHL’1ı hastaların hepsinde standart başlangıç tedavisi bir antrasiklin içeren çoklu ilaç kemoterapi rejimidir. En yaygın kullanılan rejim siklofosfamid, doksorubicin, vincristine ve prednizondan oluşan (CHOP) kombinasyondur. Bu tedavi rejimi m-BACOD, Pro-MACE ve MACOP-B gibi daha karmaşık ve daha yoğun rejimIere denk gibi görünmektedir ve bu yüzden CHOP standart tedavi olarak kabul edilmektedir. Erken evre olan hastalar (I veya büyük kitleli olmayan evre II) eğer kemoterapi kullanımı sınırlıysa en azından üç evre CHOP uygulamasında sonra bölgesel radyoterapi uygulanmalıdır. İleri evre olanlarda altı defa CHOP kullanımı gerekmektedir; ileri evre hastalığın büyük kitleli formunda bölgesel radyoterapinin rolü ortaya konamamıştır. CHOP veya benzer rejimlerle tam remisyon başanlabilmektedir, hastaların %30-40’ında kül’ sağlanabilmektedir. Kötü prognozlu seçilmiş hastalar yüksek doz kemoterapi ve ilk remisyonda uygulanacak otolog kök hücre desteğinden yarar görebilirler.
Tek başına CHOP kemoterapisi ile karşılaştırıldığında, başlangıç CHOP kemoterapisine Rituksimab ilavesinin yaygın B-hücreli lenfomalı yaşlı hastalarda yanıt hızını arttırdığı ve sağ kalımı uzattığı gösterilmiştir. Bu yüzden CHOP kemoterapisi ile kombinasyon olarak rituksimab, yaygın B-hücreli lenfomalarda başlangıç tedavisi şeklinde günümüzde yaygın olarak kullanılmaktadır. Remisyon sağlandıktan sonra relaps gösteren hastalar, eğer relaps gösteren hastalık standart doz kemoterapiye hala yanıt veriyorsa, otolog periferik kan kök hücre desteği veya transplantasyonuyla yüksek doz kemoterapi kullanılarak genellikle tedavi edilebilmektedir. Koloni stimülan faktör (CSF) desteğiyle periferik kan kök hücreleri kullanılarak bu prosedür sayesinde 1990’dan bu yana morbidite ve mortalitede bir azalma sağlanmıştır. Bu prosedür ciddi ko-morbiditesi olmayan hastalarda güvenle kullanılabilmektedir. Otolog kök hücre transplantasyonu ile yüksek doz kemoterapi, standart doz salvage (kurtarma) rejim uygulamasından daha üstündür ve relaps gösteren kemosensitif yaygın agresif NHL hastalarında standart tedavi olarak kabul edilmektedir.Mantle Cell (Hücreli) Lenfoma NHL’ların sınıflandırılmasında immünfenotiplendirme standart uygulama haline geldiğinden bu yana mantle cell (hücreli) lenfomaların tanısı artan sıklıkla konmakta ve REAL/WHO sınıflandırmasında yer almaktadır. Mantle cell lenfoma Uluslararası Çalışma Formülasyonunda (ÇF) tanımlanmıştı ve ÇF’da genellikle yaygın bölünmüş hücreli veya yaygın mikst (karma) hücreli lenfoma olarak tanı almaktaydı. Bütün NHL’ların %5-8’ini oluşturmaktadır. Mantle cell lenfomalar, lenfoid folikülün mantle bölgesinden geliştiği düşünülen olgun B-hücreli neoplazmalardır. Karakteristik immünfenotip özellikleri vardır. Mantle hücreleri, olgun B-hücre belirteçIeri (CD19, CD20 ve CD21) ile birlikte CDS antijeni taşırlar, fakat tipik olarak CD23 ekspresyonu göstermezler. Mantle cell lenfomalar, patolojik ve klinik olarak KLL (ki CDS pozitif olan bir tek diğer NHL’dir) ile kolaylıkla karıştırılabildiğinden, CD23 yokluğu mantle cell lenfomayı KLL’den (ki CD23 pozitiftir) ayırt etmekte önemli bir özelliktir. Mantle cell lenfomalar, siklin DI proteini (growth-promoting protein cyclin D i) kodlayan BeLi veya PRADJ geni ile immünglobulin ağır zincirini yan yana sıralayan patolojik olarak tanı koydurucu t( 1 1;14) translokasyonuyla karakterizedir. Mantle cell lenfomalar, slkJıkla kemik iliği tutulumu olan ileri evre hastalardaki agresif olmayan lenfonulara genellikle bir çok yönden benzerlik gösterirler. Bu lenfomalar özellikle Waldeyer halkası ve GİS’i tutmaya eğilimleri bulunur. Düşük gradeli foliküler lenfomalarda olduğu gibi, mantle cell lenfomalar tedavi edilebilir, fakat kür sağlanamaz. Ancak, nonagresif lenfomaların aksine, bu neoplazmalar biyolojik olarak agresiftir ve sadece 2-3 yıllık medyan sağ kalım gösterir. Foliküler lenfomalarla karşılaştırıldıklarında daha agresif doğasından dolayı mantle cell lenfoma hastaları tanı konar konmaz sistemik kemoterapi ile tedavi edilir, fakat uzun süreli remisyon sağlanaması zordur. Bu zorlayıcı alt grup için optimal bir tedavi henüz oluşturularnamıştır ve bu hastalarda immünterapi veya transplantasyon dahil deneysel terapilerin uygulanması düşünülmelidir.
Yüksek grade NHL’nin iki alt grubu; Burkitt veya küçük bölünmemiş hücreli ve lenfoblastik lenfomadır. Erişkin nüfusta oldukça nadir görülmektedir. Bununla beraber önemlidirler, çünkü uygun tedaviyle potansiyel olarak kür edilebilirler ve sıklıkla tanı konduklarında agresif doğaları ve hızlı büyüme özellikleri ve tedaviye başlandıktan sonra tümör lizis geliştirme eğilimleri nedeniyle hemen hastaneye yatırılarak tedavi edilmeleri gerekir. Lenfoblastik lenfomalar agresif lenfomalardır, T-hücreli akut lenfositik lösemiyle yakından ilişkilidir ve T-hücre immünfenotipi ve terminal deoksinükleotid transferaz bulunması özellikleriyle bir çok NHL’lardan ayırt edilirler. Genç erişkin erkekJeri etkiler, mediastinumu ve kemik iliğini tutar, leptomeninkslerde relaps göstermek eğilimindedir. Burkitt veya küçük bölünmemiş hücreli lenfoma erişkinlerde ender görülen B-hücreli lenfomadır ve oldukça agresiftir. Kemik iliğini ve merkezi sinir sistemini tutar. Burkitt lenfoma sitogenetik olarak, 19 lokusünü myc onkogeniyle yan yana sıralayan t(8; 14) translokasyonuyla karakterizedir. Çocuklarda Burkitt lenfomanın endemik olduğu Merkezi-Afrika’da sıklıkJa EBV (ebstein barr virüsü) ile ilişkilidir. Ancak, Amerika Birleşik Devletleri’nde sporadik Burkitt lenfomanın EBV ile ilişkili olması ender bir durumdur. Burkitt lenfoma ve lenfoblastik lenfoma leptomeningeal relapsı engellemek için intratekal kemoterapiyi de içeren yoğun çoklu kemoterapi uygulamasını gerektirirler. Kemoterapinin başlamasıyla bu tümörler hızla tümör Iizisi geliştirirler. Bütün hastalar tümör lizis sendromuna karşı kemoterapi öncesi ve esnasında profilaksi gerektirider. Profilaksi hidrasyon, idrar alkalinizasyonu ve allopurinolü içerir.
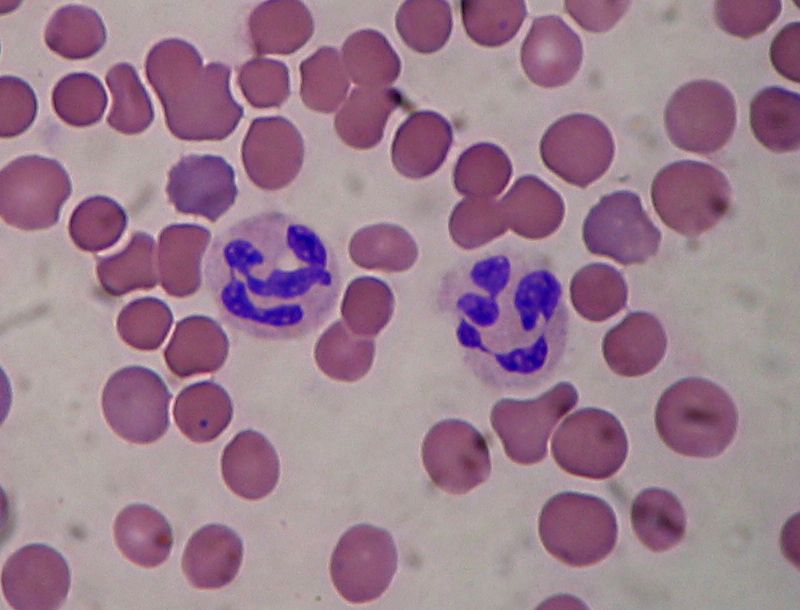
HH genç erişkinlerde en sık görülen lenfomadır. ABD ve diğer endüstrileşmiş ülkelerde bimodal yaş dağılımı gösterir: 15-35 yaş arasında daha geniş bir dağılım zirvesi, ikinci olarak daha küçük dağılım zirvesini 50 yaşından büyük hastalarda yapar. HH nedeni hala bulunamamıştır. Malign HH hücrelerinde EBV sıklıkla mevcut olmakla birlikte, EBV ile HH arasında doğrudan bir sebep sonuç ilişkisi saptanamamıştır. HH konjenital immün yetmezlik sendromlu hastalarda veya immünitesi baskılanmış organ nakli alıcısı hastalarda yüksek sıklıkta görülmemektedir. HIV pozitif hastalarda HH riski arttığı gösterilmemiştir. HH tanısı tutulan lenf düğümünde Reed-Sternberg (RS) hücresinin tanılanmasıyla yapılır. Klasik RS hücresi büyük olup çift nükleusu vardır. Her nükJeusunda belirgin nükleolusu olmasından dolayı çekirdeğine baykuş gözü görünümü vermektedir. RS hücresinin hücresel kökeni on yıllarca tartışmalı kalmıştır. Ancak, moleküler çalışmalarda germ dizisi 19 lokusünün klonal rearanjmanı olduğunu göstermiştir ki RS hücresi, sitoplazmasının veya hücre yüzey immünglobulinleri olmamasına karşın, B hücresi doğasında olduğu saptanmıştır. NHL ve diğer malign neoplazmaların aksine, HH tutulan lenf düğümündeki infiltrat kitlesi sıklıkla benign reaktif enflamatuar hücrelerden oluşmaktadır. Tanı koydurucu RS hücresini bulmak genellikle zordur. İmmünfenotiplendirmede klasik RS hüci’esi CD30 (Ki-I) ve CD 15 pozitif ve CD 20, CD45 ve sitoplazmik veya yüzey immünglobulinleri için negatiftir. HH’lı olguların yaklaşık yarısında RS hücrelerinde EBV taııılanmıştır. Hodgkin hastalığının dört patolojik tipi tanımlanmıştır. Nodüler sklerozan tip (NS) en sık olarak karşılaşılanıdır (HH’lilerin yaklaşık %80’inde) ve lenf düğümünü nodüllere ayıran fibröz bantlar ve laküner tip RS hücrelerin olmasıyla karakterizedir. Adölesanlarda ağırlıkla görülen bu form tipik olarak mediastinumu ve diğer supradiafragmatik bölge lenf düğümü alanlarını tutar. Mikst hücreli (MC) tip HH’li olguların ortalama % 15’ine oluşturur, bantlar oluşturan skleroz yoktur ve RS hücreleri yaygın infiltrasyon alanı (ki NS tipteki infiltratla karşılaştırıldığında daha heterojendir) içinde kolaylıkJa tanılanabilir. MC tip herhangi bir yaş grubunda görülebilir ve subdiafragmatik tutulumlu ileri evre hastalık MC tip HH’da NS tipteki HH’na kıyasla daha çok görülmektedir. Lenfositten fakir tip nadirdir, HH’1i olguların %1 'den azını oluşturur.
Enflamatuar hücreler azalmasına karşın RS hücresi yaygınlığıyla karakterizedir. Lenfositten fakir tip yaşlı erişkinler, HIV pozitif hastalar ve en düstrileşmesini tamamlamamış ülke insanlarında daha yaygındır. Lenfositten zengin (lymphocyte-predominant, LP) tip ayrı bir konu olarak ortaya çıkmaktadır: gerçek bir HH olarak ele alınmaktadır ancak nonagresif NHL’yla HH ile olduğundan daha fazla yakından ilişkilidir. RS hücrelerin değişik bir çeşidi ile beraber nodüler büyüme düzeni göstermesiyle karakterizedir. Buradaki RS hücrelerin birden fazla loblu nükleusları vardır ve bu hücrelere ‘patlamış mısır’ hücreleri (popcom cells) denir; klasik RS hücreleri genellikle görülmez. Bu atipik hücrelerin immünfenotiplendirmesi klasik RS hücrelerinkinden farklıdır: B-hücre antijenleri (CDI9 ve CD20) ve CD45 ekspresyonu mevcuttur, fakat klasik RS belirteçleri olan CD15 ve CD30 yoktur. HH2nın yaklaşık %5’ini oluşturmaktadır. Erkeklerde daha sıktıL Periferik lenf düğümlerini tutma eğilimi gösterirken, mediastinumu tutmaz. Geç dönem relapsları, diğer HH tiplerinden daha sık olmasına karşın, prognoz mükemmeldir. HH en sık mediasten veya servikal lenf düğümlerinden çıkar. Bunların komşuluğundaki bitişik veya bitişik olmayan, retroperitoneal ve dalak dahil, lenf düğüm lerine yayılır. Hastalık ilerledikçe, kemik iliği, dalak, ve akciğer dahil ekstranodal alanları tutmak üzere hematojen yolla yayılır.
Lenf düğümünden hematojen yolla yayılarak bitişiğindeki ekstranodal alanları tutmasına karşın (örneğin bitişiğindeki lenf düğümlerinden vertebraları tutar, hiler lenf düğümlerinden akciğer parankimine yayılır), NHL’ların aksine, HH ekstranodal alanlardan nadiren çıkar. En sık servikal olmak üzere, HH genellikle ağrısız lenfadenopati görülür. Asemptomatik hastalarda rutin göğüs filminde mediastinal lenfadenopati kazara fark edilebilir. Eşlik eden pulmoner tutulum veya pulmoner tutulum olmaksızın büyük hilar veya mediastinal lenfadenopati öksürük, nefes darlığı, wheezing veya stridor gibi solunumsal yakınmalara yol açabilir. Hodgkin hastalarının yaklaşık üçte birinde ateş, gece terlemesi, kilo kaybı (B semptomları) vardır ve bunlar ilk yakınmalar olarak karşımıza çıkabilir. B semptomları na ek olarak, yaygın kaşınma da HH ile birlikte görülebilir ve NS tip ile korelasyon gösterir. Hastalar, öykülerinde HH tanısı konmadan önce aylar hatta yıllardır varolan, geçmeyen kaşınma yakınması verebilir. HH, intradermal deri testlerine aneıjik yanıt gösteren işlevsel T-hücre bozukluğu ile bağlantılı olmasına karşın, fırsatçı enfeksiyonlar nadiren görülmektedir. Sıklıkla yavaş seyirli olsa da tedavi edilmediği taktirde, hematojen yolla kemik iliği, karaciğer ve diğer organlara yayılımın gösterdiği ve ani bir şekilde birden fazla lenf düğümü tutuğu görülmektedir. Hastalık ilerledikçe, B semptomları, bitkinlik, kaşeksi ve enfeksiyonlar görülür ve ilerlemiş Hodgkin hastalıklı olgular kemik iliği yetmezliği veya enfeksiyonlara bağlı komplikasyonlardan dolayı ölürler. Yeni tanı alan Hodgkin hastalarının doğru evrelenmesi; tedavi planlaması, prognoz ve tedaviye yanıtın öngörülebilmesi açısından önemlidir. Modifiye Ann Arbor sınıflaması kullanılmaktadır. A veya B son ekleri ateş, gece terlemesi veya kilo kaybı semptomlaımın olması (B) veya olmamasını (A) gösterir. Yeni tanı almış Hodgkin olguIarının evreleme çalışması NHL’lı hastalarınkine benzemektedir. Hastalar iyi bir öykü ve fizik bakıdan geçirilmelidir: tam kan sayımı, eritrosit sedimentasyon hızı, göğüs filmi, toraks, abdomen ve pelvis bilgisayarlı tomografi incelemeleri, kemik iliği aspirasyon ve biyopsisi ve PET veya galyum taraması yapılmalıdır. Subdiafragmatik lenfadenopatilerin değerlendirilmesinde lenfanjiyografinin tarihsel önemi vardır, ancak bu testin yapılması ve değerlendirilmesinde gerekli olan uzmanlık artık yaygın olarak bulunmadığından bunun yerine CT taraması ve nükleer görüntüleme tekniklerinin kombinasyonları tercih edilmektedir. Kemik filmleri, kemik taraması ve spinal MR görüntülernesi gibi ek çalışmalar sadece bu yapıların tutulumunu düşündürecek semptomların varlığında gerçekleşti ri lmelidir. Bu girişimsel olmayan çalışmalardan elde edilecek bilgi Hodgkin hastalıklı olgunun klinik evrelemesini yapmanuzı sağlar. Erken evre hastalık tedavisi gelişmesiyle birlikte, diafragma altı tutulumu daha iyi belirlemek için yapılan evreleme laparatomisi işlemi 1990’lardan bu yana gittikçe azalma göstermiştir. Bu prosedür laparatomi ile birlikte splenektomi, karaciğer biyopsisi ve retropritonal lenf düğümlerinden örnekleme yapılmasını gerektir. Bu işlemden elde edilen bilgi hastalığın patolojik evresini tanımlar.

Primer radyoterapinin uygulanması sayesinde tedavinin gelişme göstermesi sonucu evreleme laparatomisi günümüzde rutin olarak yapılmamaktadır (daha sonra tartışılacaktır). Ancak, eğer supradiafragmatik evre 1 veya 5 hastalığı olan bir olgu tek tedavi yöntemi olarak sadece radyoterapi alacaksa, dalağın ve retroperitoneal lenf düğümlerinin henüz ortaya çıkmamış gizli bir tutulumunun olmadığını gösterebilmek için evreleme laparatomisi yapmak gereklidir. Gizli HH, klinik evre 1 veya 5 hastaların %30 kadarında diyafram altı yerleşimli bulunabilir ve kemoterapi uygulamasını gerektirir. Splenektomi ile birlikte evreleme laparatomisi yapılan hastaların kapsüııü mjkroorganizmaların neden olduğu enfeksiyon riski vardır. Bu yüzden pnömokok ve Haemoplıilus injluenzae aşılarının aıneliyattan önce yapılması gerekir. HH’da relaps veya sağ kalım risklerini etkileyen bir grup prognoz belirleyici faktörler tanımlannuştır. En önemli kötü prognoz belirleyici faktörler: MC veya lenfositten fakir tip histolojiler, erkek cinsiyet, yüksek sayıda lenf düğümü alanlarının tutulması, ileri yaş (> 40 yaş üzeri), B semptomlarının varlığı, yüksek eritrosit sedimantasyon hızı ve hastalık kitle yükünün fazla oluşu (mediastinumun üçte birden fazla genişlemesi veya 10 cm’den geniş ebatta lenf düğümü kitlesinin
oluşu). Erken evre olgularda bu etmenlerden herhangi birinin oluşu, gizli abdominal tutulum veya primer radyasyon tedavisinden sonra relaps riskinj arttırır ve böylece başlangıç tedavisine kemoterapinin eklenmesi kararını etkiler. HH tedavisi 1980’den bu yana belirgin olarak gelişme gösterdi. HH kür elde edilebilir; günümüz tedavi modaliteleri kullanılarak kür oranı %80’i geçmektedir. HH’lı olguların çoğu genç erişkinlerdir ve uzun süreli hastalıksız sağ kalım süreleri vardır, küratif potansiyeli feda etmeksizin tedaviye bağlı morbiditeyi ve mortaliteyi en aza indirecek tedavilerin uygulanması vurgulanmaya çalışılmaktadır. Tutulan bölgeler ve yanındaki lenf düğümü alanlarına ılımlı dozlaı-da radyasyon tedavisi (>35 Gy) düşük riskli, erken evre (ters (advers) risk faktörleri olmaksızın büyük kitleli olmayan evre 1 ve 5A) taşıyan
olguların çoğunda küratiftir. Bu hastalar için yaşam sağlayıcı tedavi seçeneği olarak devam etmektedir. Ancak standart dozlarda radyasyon tedavisi alan hastaların uzun dönem izlemi radyasyon verilen bölge içinde ve kenarlarında on yıldan fazla bir süre sonra bir takım solid tümör oluşumu riskinin artmış olduğu sonucunu göstermiştir.
HH’da toraks radyasyonu, kadınlarda meme kanseri, erkeklerde ve kadınlarda akciğer kanseri riskinin özellikle artış göstermesiyle bağlantılıdır. Ek olarak HH’da standart radyasyon tedavisinin uzun dönem sekellerini tiroit disfoksiyonu (sıklıkla hipotiroidizm) ve koroner arter hastalığında artış oluşturmaktadır. Bu yüzden düşük riskli erken evre HH’da standart dozlarda primer radyasyon tedavisi uygulama isteği toraks radyasyonu gerektiren hastalarda, ki bunlar erken evredeki hastaların büyük bir çoğunluğunu teşkil eder, azalmaktadır. Standart doz radyasyonun uzun dönem karsinojenik etkilerinin tanınmasına yanıt olmak üzere, düşük riskli erken evre HH olan olguların tedavi yaklaşımı geliştirilmektedir. Kemoterapi (örneğin, doksorubisin [Adriamisin], bleomicin, vinblastin, ve dacarbazin, [ABVD] rejimi olarak adlandırılır) ile düşük doz radyoterapi (>30 Gy) kombinasyonu tercihi gittikçe artan bir uygulamadır. Bu kombinasyonun ikincil solid tümör gelişme riski ile ilişkisi olmamaktadır. Erken evre HH’da düşük doz radyasyon ile kemoterapi kombinasyonu uygulamasının optimal süresi henüz tam belirlenmemiştir. ileri evre Hodgkin hastalığı (evre III veya IV) veya kötü prognoz işaretleri olan (örneğin büyük kitleli, B semptomları olması, MC tipte histoloji) erken evre olgular yüksek relaps hızı nedeniyle tek tedavi modalitesi olarak radyasyon terapisine uygun adaylar değildirler. Bu hastalar kemoterapi ile tedavi edilmelidirler. Nitrojen mustard, vincristine (Oncovin), procarbazin ve prednizon (MOPP olarak adlandırılır) içeren çoklu kemoterapi programlfun 1970’lerde ileri evre olgularda yüksek oranda küratif olduğu gösterilmişti. MOPP hala yüksek oranda küratiftir, ancak uzun dönem toksik etkileri nedeniyle güncel uygulamada nadiren kullanılmaktadır. Bu uzun dönem toksik etkiler, MOPP alan erkeklerin yaklaşık tamamında sterilite ve kadınların önemli bir kısmında infertilite olmak üzere, yüksek oranda akut miyeloid lösemi geliştirme riskidir. ABVD rejimi günümüzde ABD’de en yaygın uygulanan protokoldür. ABVD rejimi MOPP kadar etkindir, fakat sterilite, infertilite veya tedaviye bağlı lösemiye neden olmaz. Bu rejimde bleomisin olması nedeniyle ABVD’ye bağlı pulmoner fibrozis düşük oranda «%5) görülmektedir. Daha önceden akciğer hastalığı olan veya tedavi protokolünün bir parçası olarak toraks bölgesine radyasyon alanlarda pulmoner fibroz geliştirme riski en yüksektir. Daha önceden kardiyomiyopatisi olan hastalarda doksorubisin bağlı daha ileri kardiyak hasar riski nedeniyle ABVD tedavisine uygun değillerdir. Bu hastalarda alternatif rejimler gereklidir.
Radyoterapinin kemoterapi ile kombinasyonu ileri evre Hodgkin hastalığı tedavisinde genellikle kullanılmamaktadır. Ancak, büyük kitleli mediasten tutulumu olan olgularda kemoterapinin tamamlanmasından sonra mediastinuma ilave radyoterapi uygulanmasının relaps hızını azalttığı gösterilmiştir. Bu yüzden kombine modalite kemoterapi (kemoterapi artı radyoterapi) büyük kitleli mediastinal hastalığı olan olgularda standart olarak kabul edilmektedir. HH’da hastanın tedaviye yanıtını değerlendirme tedavi süresinde ve bitiminde evreleme değerlendirmesinin tekrar edilmesini gerektirir (fizik inceleme, CT, galyum veya PET tarama ve eğer tanı aldığında pozitif idiyse kemik iliği biyopsisi). Göğüs filminde veya CT’de devam eden radyografik anormallik olmasına karşın (örneğin büyümüş lenf düğümleri, arta kalan mediasten kitlesi) hastalarda kür sağlanabilir. Tedaviye başlangıç yanıtından sonra arta kalan radyografik anormallik olan hastalar süregelen HH’na ait ilave teyit edici bulgu olmaksızın (örneğin biyopsi ile doğrulama veya belli bir zaman geçtikten sonra radyografik doğrulama gibi) salvage terapiye tabi tutulmamalıdır. Arta kalan radyografik anormalliği olan hastalardaki inatçı pozitif galyum veya PET taraması, yüksek nüks hızıyla bağlantılıdır. Bu hastalar yalandan izlenmelidir veya öncelikli yineleme biyopsisi ve/veya salvage terapisi açısından değerlendirilmelidir. Ne yapılırsa yapılsın relaps gösterecek olan hastalar 2 yıl içinde nüks göstereceklerdir; 5 yıldan sonraki nüksler son derecede enderdir. Nüks eden veya başlangıç tedavisine yanıt vermekte başarısız olan hastalara salvage tedavisi uygulanmalıdır. Eğer uygun tedavi almışlarsa bu olguların çoğunda günümüzde kür sağlanabilir. Standart dozda radyoterapi alan (kemoterapi olmaksızın) erken evre HH’larının yaklaşık %20’si nüks gösterir. Bu hastalar standart kemoterapi ile (örneğin ABVD) kurtarılabilmektedir. Standart kemoterapiden sonra nüks gösteren hastalar otolog periferik kök hücre desteği ile birlikte yüksek doz kemoterapi verilerek tedavi edilmelidirler. Nüks gösteren kemoterapiye duyarlı Hodgkinli hastaların %5O’sinden fazlası bu rejimle kür edilebilirler.
B-hücreli KLL, B-hücre kökenli küçük lenfositlerin sayıca çoğalması ve birikimi ile karakterize malign lenfosit bozukluğudur. REAL ve ÇF sınıflamasında tanımlanan B-hücreli küçük lenfositik lenfoma ile özellikle özdeştir. ABD’de en yaygın görülen lösemi tipidir. Erkeklerde kadınlardan iki kat daha fazla görülür. Yaşamın herhangi bir evresinde görülebilmesine karşın, yaşla birlikte insidansı artar, olguların %90’dan fazlası 50 yaştan yukarı erişkinlerde tanılanır. Nedeni bilinmemektedir. Hastalık için yatkınlık oluşturacak belirgin bir genetik temel bulunamamıştır. çevresel etmenler, örneğin radyasyon etkisi ve karsinojenlere maruziyet kanıtlanamamıştır. KLL’nin yaygın formu olgun B-hücrelerin klonal çoğalmasıdır. Karakteristik olgun B-hücre belirteçleri ve tümörün klonal kökenli olduğunu gösteren düşük düzeyde hafif zincir kısıtlı yüzey M-immünglobulinleri (lgM) vardır. Ek olarak KLL B-hücreleri, normal B-hücrelerinin küçük alt yapısını işaret eden CDS molekülü ve CD23 (immüngloulin E [lgE] için Fe reseptörü) taşırlar. Dolayısıyla KLL’nin tanı koydurucu irnmünfenotiplendirmesinde olgun B-hücre topluluğu olduğunu gösteren, hafif zincir kısıtlı veya 19gen düzenleme çalışmaları sonucunda, karakteristik B-hücre belirteçieri (CD19,CD20, ve CD21) taşıdıkları ve CDS ve CD23 için pozitif oldukları gösterilmiştir. KLL’de patognomonik kromozomal anormallik tanımlanmamışsa da hastaların %30-50’sinde sitogenetik anormallikler vardır. En sık görülen kromozomal anormallikler kromozom 12 (sıklıkla trizomi 12), 13 veya 14’ü içerir ve bu sitogenetik anormalliklerin varlığı kötü prognoz göstergesidir. Kemik iliği veya periferik kan yayması nukleolusları fark edilemeyen küçük lenfositlerin hakimiyetini yansıtIJ ve tutulan lenf düğümlerinde normal yapıyı bozan bu hücrelerin yaygın infiltrasyonu görülür.KLL hücrelerinin kemik iliği, periferik kan, lenf düğümleri ve dalakta birikimi sonucunda lenfositoz, kemik iliği işlevlerinde azalma, lenfadenopati ve splenomegali gelişir. KLL’de ayrıca immün sistem düzenlenmesinin bozulmasıyla sıklıkla karşılaşılır. Bakteriyel enfeksiyon gelişim riskini arttıran hipogammaglobulinemi ve coombs pozitif hemolitik anemi veya immün trombositopeni gibi otoimmün olaylar görülür. Tanı rutin tam kan sayımında tesadüfen konulur: Küçük lenfosit hakimiyeti ile birlikte lökositozu gösterir; periferik kanın flow sitometrik analizi veya kemik iliği aspirasyonunda karakteristik klonal CD5 ve CD23 pozitif B-hücre topluluğu saptanır. Bazı hastaların lenfadenopati, sitopeniye bağlı semptomlar veya yineleyen enfeksiyonları olur. Hastalık ilerledikçe yaygın lenfadenopati, hepatosplenomegali ve kemik iliği yetmezliği gelişir. Tedaviye yanıt vermeyen hastalarda ölüm sıklıkla yineleyen enfeksiyonlardan veya kemik iliği yetmezliğinden kaynaklanır. Olguların yaklaşık %5’inde, KLL oldukça malign yaygın büyük hücreli lenfomaya dönüşür, sıklıkla hızlı bir şekilde ölümle sonuçlanır; bu dönüşüm yaygın olarak Richter sendromu olarak adlandırılır. KLL düşük dereceli lösemi veya lenfomadır. Tipik olarak yılları hatta on yılları aşan bir süreci olan yavaş ilerleme gösteren uzun doğal öyküsüyle karakterizedir. Medyan sağkalım süresi 6 yılın üzerindedir. Tanı konduğunda hastalığın yaygınlığı veya evresi sağkalım için en iyi öngördürücüdür.
KLL için en yaygın kullanılan Rai ve Binet evreleme sistemleri görülmektedir; hastaların çoğunda, evre O, i veya II hastalık vardır. Standart tedavinin küratif olmaması ve KLL’nin yıllar süren uzun bir asemptomatik dönemi olması nedenlerinden, özgün tedaviye başlanması için hasta semptom (örneğin, büyük kitleli lenfadenopati, hastalığı düşündürecek yapısal semptomlar; ateş gibi, kemik iliği infiltrasyonundan veya otoimmün olaydan dolayı sitopeni) geliştirinceye kadar beklenebilir. Tedavi gerektiği zaman, başlangıç terapi si ya bir alkilleyici ajan örneğin klorambusilin prednizonla kombinasyonu veya nükleosit-analoğu olan fludarabin ile başlatılabilir. Hastaların çoğu bu girişimlerden herhangi birine tümör hasarında belirgin bir azalmayla yanıt verir. Klorambusil terapisiyle karşılaştırıldığında fludarabin tedavisi daha yüksek oranda tam remisyon sağlamaktadır ve kombine rejimIerin (örn., fludarabin, siklofosfamit ve rituksimab) özellikle güven verici sonuçlar vermekte olduğu gösterilmiştir. Yineleyen veya tedaviye yanıt vermeyen hastalık alemtuzumab (pek çok lenfosit üzerinde bulunan CD52 molekülüne karşı geliştirilmiş insan monoklonal antikoru), tedavisine yanıt verebilir. Rituksimab da yineleyen KLL olgularında aktif bir ajandır. Otoimmün olay geliştiren hastalar kortikosteroidlerle tedavi gerektirir. Hipogammaglobulinemi gelişen hastalardaki enfeksiyon sıklığını azaltmak için intravenöz gammaglobulin kullanılabilir. Hızla büyüyen mediastinal kitle, yapısal semptomların gelişmesi ve yüksek LDH düzeylerinin ortaya çıkması hastalığın kötü prognoz gösteren yaygın büyük hücreli lenfomaya dönüştüğünü düşündürür.
Saçaklı hücreli lösemi (SHL, tüylü hücreli lösemi, hairy cell leukemia) morfolojik olarak saçaklı (hairy) karakteristik görünümü olan neplastik B hücrelerinin kemik iliği, periferik kan ve dalakta birikimiyle karakterize, biyolojik olarak yavaş seyirli neoplastik lenfoid bozukluktur. Saçaklı hücreler ince sitoplazmik çıkıntıları olan lenfoid hücrelerdir. Bu hücreler tartarata dirençli asit fosfataz aktiviteleri, B-hücre immünfenotipinde oluşları ve hafif ve ağır zincir immünglobulin genlerinin yeniden düzenlenmesiyle doğrudan ayırt edilirler. Periferik kanda veya kemik iliği biyopsisinde tipik saçaklı hücrelerin gösterilmesiyle tanı konur. Tipik olarak yoğun retikülin fibrozisin varlığı nedeniyle kemik iliğinden aspirasyonla genellikle materyal elde edilememesi hastalığın diğer bir özelliğidir. Bu hastalık yüzeysel olarak KLL’ye benzer. Fakat
kendine özgü klinik özellikleri vardır ve farklı terapi uygulanmasını gerektirir. Saçaklı hücreli lösemili asemptomatik bir hastada rutin tam kan sayımıyla tanı konabilir; asemptomatik hastalar daha sonra genellikle splenomegali, azalmış konak savunmasına bağlı enfeksiyonlar veya vaskülit/artrit gibi eşlik eden otoimmün sendromIara atfedilebilecek semptomlar gösterebilirler. Osteolitik kemik lezyonları oluşabilir ve ağrı ya neden olabilir. B semptomları nadirdir. Fizik muayenede, splenomegali hastaların %80’inden fazlasında mevcuttur; hepatomegali daha az sıklıktadır ve lenfadenopati ayırt edici bir özellik olarak nadirdir. Tanı konulduğunda pansitopeni tipik olarak vardır. SHL seyri genellikle sakindir, yavaş gelişen pansitopeni ve splenomegali görülür. Ancak hastalığın ilerleme hızında ve ciddiyetinde belirgin değişkenlikler bulunabilir. Etkin terapiden önce bakteriyel ve fungal enfeksiyonlar sıklıkla gelişiyor ve başlıca ölüm nedenini oluşturuyordu. Belirgin sitopeni veya hastalığın diğer komplikasyonları
olmayan asemptomatik hastalar, acil terapi gerektirmezler, hastalığın ilerlemesi veya enfeksiyöz komplikasyonlar açısından yakın izlem altında tutulabilirler. Orta derecede sitopeni, enfeksiyöz komplikasyonlar, hızlı seyirli hastalık, semptomatik splenomegali, kemik tutulumu veya otoimmün sendromları gösteren olgular terapi almalıdır. Birinci sıra terapi purin nüklosit analogu olan 2-klorodeoksiadenozi ndir.
Plazma hücresi bozuklukları veya diskrazileri, monoklonal immünglobulin (veya immünglobulin parçası) veya M proteini üretimi özellikleri nedeniyle birbirlerine bağlı bir grup B-hücresi neoplazmalarından oluşur. Bu bozukluklardaki tümör hücreleri yüksek hızda immünglobulin üretmeye uyarlanmış farklılaşmış plazma hücresi özelliklerini gösterir. Protein elektroforezi ile idrar ve serumda gösterilebilen homojen bir immünglobulin molekülü veya immünglobulin molekül parçasının varlığı ayırt edici laboratuar testidir. İliğin in infiltrasyonunun doğrudan etkileri kadar M proteinin sistemik etkileri de genellikle görülür. Plazma hücresi diskrazileri sınıflandırması, kısmen, üretilen immünglobulin (immünglobulin G [IgG], immünglobulin A [IgAL, immünglobulin D, IgE veya IgM) veya immünglobulin parçası (ağır zincir veya hafif zincir) tarafından belirlenir. En yaygın plazma hücresi neoplazmaları multipl myelom (multiple myeloma) ve buna yakından ilişkili plazmasitoma (kemik ve ekstramedüller yumuşak dokunun ayrı bir tümörüdür); diğer daha az yaygın plazma hücresi neoplazmaları olan Waldenström makroglobulinemisi, ağır zincir hastalığı ve primer amiloidozdur. M proteini plazma hücresi diskrazileri dışındaki benign ve malign durumlarda da bulunabilir. KLL’li hastaların yaklaşık % 10’unun serumunda monoklonal immünglobulin G veya M gösterilebilir düzeylerde bulunur. M proteinleri bir grup otoreaktif veya enfeksiyöz bozukluklarda bulunabilir. Ek olarak, eşlik eden belirli bir hastalığı olmayan veya plazma hücresi bozukluklarına bağlı herhangi bir klinik bulgu veya laboratuar bulgusu olmayan bireylerin serum protein elektroforezinde M proteini bulunabilir. Bu bulgu önemi bilinmeyen monoklonal gamopati (monodonal gammopatlıy of unknown significance, MGUS) olarak adlandırılır ve düşük düzeylerde serum M proteini varlığı «3 g/dL), idrar Bence Jones proteini yokluğu, kemik iliğinde % 10’dan az plazma hücresi varlığı, anemi olmayışı, hiperkalsemi, renal yetmezlik ve litik kemik lezyonların varlığı ile tanılanır. MGUS miyelomdan daha yaygındır ve yaşlanmayla sıklığı artar. Elli yaşından yukarı nüfusun % 1-2’sinde görülür. MGUS pre-malign bir durum olarak değerlendirilir. Genel nüfusla karşılaştırıldığında klinik miyelom veya malign plazma hücresi neoplazması geliştirme riski MGUS olgularında yedi kat daha yüksektir. Bununla birlikte, her yıl sadece yaklaşık %i hastada MGUS’tan klinik plazma hücresi neoplazmasına dönüşüm olmaktadır. Kararlı, ilerleme göstermeyen MGUS hastalarını sonunda gerçekten miyelom geliştirecek olanlardan ayırt etmek zordur. IgA veya IgM M proteinleri olan hastalarda ve M proteini başlangıç konsantrasyonları 1.5 g/dL üzerinde olan olgularda hastalığın ilerleme gösterme riski yüksektir. MGUS tanısı olan hastaların izlenmesinin sağ kalımı arttırdığına dair kesin kanıtlamamakla birlikte, belirgin semptomlar veya komplikasyonlar ortaya çıkmadan önce multipl miyeloma ilerleme olup olmadığını değerlendirmek için, serum elektroforezi dahil olmak üzere, hastalar yıllık değerlendirmeye tabi tutulmalıdır.

Multipl miyelom malign plazma hücresi bozukluğudur. Kemik ve kemik iliğinin neoplastik infiltrasyonu ve serum veya idrarda monoklonal immünglobulin veya hafif zincirlerin varlığı ile karakterizedir. Kemik iliğinde plazma hücre sayısında artış (%30’dan fazla) ve 19 M’den ayrı olarak serum M proteinlerinde artış (IgG > 3g/dL, IgA > 2 g/dL) veya idrar M protein miktarının 1g124 h’den fazla oluşu ile tanılandırır. Düşük düzeylerde M proteini olan veya kemik iliğinde %30’dan az plazma hücresi olan hastalar hipogammaglobulinemi, litik kemik lezyonlar veya plazmasitoma gibi diğer özelliklerin bir arada olmasına dayanılarak da miyelom tanılanabilir. Bu özellikleri olmayan hastalar için, asıl ayrıcı tanı MGUS ile miyelom arasındadır; bazı olgularda, sadece bir dizi izlem yapılarak M proteini düzeylerinde yükselme kanıtları veya miyelom klinik bulguları gelişiminin değerlendirilmesi ile ayrım yapılabilir. Yaklaşık MM’lı hastaların %20’sinin serumunda standart elektroforezle gösterilebilir miktarlarda M proteini yoktur. Fakat idrar protein elektroforezinde serbest hafif zincirleri (Bence Jones protein) 24 saatlik idrarda ölçülebilen miktarlarda saptanabilir (hafif-zincir hastalığı). Nadir görülen salgılaması olmayan miyelom (nonsecretory miyeloma) hastalarında ölçülebilir miktarlarda serum veya idrar M proteini yoktur. Ancak, bu hastalarda, sitoplazmik hafif zincir kısıtlı immünglobulinin immünhistokimyasal yöntemle gösterilmesi ile plazma hücrelerinin monoklonal bir grubu gösterilebilir.
Son dönemlerde, MM’lı hastaların serumlarında serbest hafif zincirlerin gösterilebilmesi için niceliksel yöntemlere ulaşılabilirlik yaygınlaşmaktadır ve hastalığı değerlendirmekte elektraforetik ölçümlere benzer bir şekilde kullanılmaktadır. Tam immünglobulin moleküllerinin haftalarla ölçülen yarılanma süreleriyle karşılaştırıldığında, dolaşımdaki serbest hafif zincirlerin görece kısa yarılanma süreleri vardır, 2-6 saat kadardır. Bu nedenle, hastaların tedaviye yanıtlarının daha hızlı olarak değerlendirilebilmesinde serbest hafif zincirler bir ölçüt olarak kullanılabilir. Kemik radyografisi tipik zımba deliği gibi tam belirgin osteolitik lezyonlar gösterir. Sıklıkla eşlik eden osteopeni ve patolojik kırıklar vardır. Kemik lezyonları spinal korda kompresyonunun eşlik ettiği çevreye bası yapan kitleler gösterebilir. Yaygın kemik tutulumunun neden olduğu hiperkalsemi miyelomda yaygındır ve klinik görünüme hakim olabilir. Kemik iliği infiltrasyonu ve hematopoezin baskılanması sonucu anemi hastaların çoğunluğunda gelişir; granülositopeni ve trombositopeni daha az görülür. Normal immünglobulinlerin üretiminin azalması ve katabolizmasının artması sonucu miyelomlu hastalar bakteriyel enfeksiyonlara yatkınlık gösterirler. Streptococcus pneumoniae, Staplıylococcus aureus, H. injluenzae ve Klebsİella pneumoniae kaynaklı solunum yolu enfeksiyonları ve gram negatif üriner sistem enfeksiyonları yaygındır. Miyelomlu hastaların yaklaşık %24’inde böbrek yetmezliği gelişir. Bu hastalarda böbrek yetmezliğinin nedeni sıklıkla çok etmenlidir; hiperkalsemi, hiperürisemi, enfeksiyon ve amiloid depolanması buna katkı yapabilir. Ancak hafif zincir üretimine bağlı doğrudan tübüler hasar her zaman vardır.
Fizikokimyasal özellikleri nedeniyle M proteinleri de olumsuz etkilerin bir diğer kaynağını oluşturabilir. Bu etkiler kriyoglobulinemi,hiperviskozite, amioloidoz ve M proteinlerinin trombositlerle ve pıhtılaşma faktörleriyle etkileşmesinden kaynaklanan pıhtılaşma bozukluklarını içerir. Miyelomda üç-tabaka lı evreleme sistemi sağkalımla korelasyon gösteren işlevsel bir sistemdir. Lenfoma ve solid tümörlerde kullanılan anatomik evreleme sistemlerinin aksine, miyelom evrelernesi tümör hasarını gösteren klinik (kemik radyografisi) ve laboratuar testlerine (hemoglobin, serum kalsiyum, serum veya idrar M protein düzeyleri ve serum kreatinin) dayanır. Kötü prognoz faktörleri ileri yaş, bozulmuş renal fonksiyon, yüksek LDH düzeyleri, anormal kemik iliği sitogenetiği ve yüksek f3ı-mikroglobulin düzeyleridir. En sonuncusu tek en güçlü sağkalım öngördürücüsüdür. Son zamanlarda, yalınlaştırılmış prognoz planlaması olarak, Miyelomda Uluslararası Evreleme Sistemi (the International Staging Sytem for Myeloma) sadece iki değişkene, mikroglobulin ve albumin düzeylerine dayanan üç evreli ayrı prognoz tanımlamıştır. Miyelomlu hastaların çoğu semptomatik, ileri evre hastalık gösterir ve terapi gerektirir. Ancak, hastaların yaklaşık %10’u evre 1 hastalık gösterir ve yavaş seyirlidir. Bu hastalar acil tedavi gerektirmezler, fakat bir dizi M protein ölçümleri ile hastalığın ilerlemesinin gösterimini gerektirirler. Yalnız kemik veya kemik iliği dışı plazmasitoması olan hastalarda lokal radyasyon terapi si uzun süreli remisyonlar sağlayabilir ve tercih edilecek tedaviyi oluşturur. Semptomatik ileri evre (II veya lll) miyelomlu hastalar destek bakım için çok titiz bir dikkat ve sistemjk tedavi gerektirirler. Miyelom kür edilebilir bir malign neoplazmasına karşın, sistemik tedavi sağ kalımı uzatabilir ve yaşam kalitesini dramatik bir şekilde arttırabiIir. Standart tedaviler kemoterapi rejimierini içerir, örneğin üçlü ilaç rejimi olarak vinkristin, doksorubicin (Adriamisin) ve deksametazon (YAD) rejimi, veya kemoterapidışı rejim olarak talidomid artı deksametazon. Talidomid İngiltere’de 1960’larda sedatif olarak kullanılmıştı, fakat gebelikte bulantıya karşı kullamldığında doğumsal anomalilere neden olduğu bulundu.
Thalidomidin anti-anjiyojenik özellikleri sonuçta kansere karşı ajan olarak geliştirilmesine yöneltti. Miyelomda talidomidin etki mekanizması açıklanamamışsa da transplantasyondan sonra yineleyen veya yanıt alınamayan hastaların üçte biri talidomide yanıt vermiştir. Kemoterapinin aksine, talidomid seyrek olarak kemjk iliğini baskılar ve periferik nöropati, kabızlık, somnolans ve döküntü gibi ayrı bir yan etki profili sergiler. Miyeloma tedavisinde başlangıç terapisi olarak talidomid ve deksametazon kombinasyonu yüksek oranda etkindir ve oral tedavi olarak kullanım üstünlüğü sağlar. Bu kombinasyonun sorun oluşturabilecek en belirgin yan etkisi hastaların %25’i kadarında görülebilen derin ven trombozudur. Diğer başlangıç sistemik terapiler tekli ilaç uygulaması olarak deksametazon veya alkjlleyici ajan temelli kombinasyon kemoterapi rejimIeridir. Ancak, alkilleyici ajan kombinasyonu kök hücre hasarına neden olur ve bu yüzden kök hücre transplantasyonuna uygun olabi lecek hastalarda kullanmaktan özellikle kaçınılır. M proteini düzeylerinde düşüşle birlikte, kemik ağrıları, hiperkalsemi ve anemide gerileme sağlanarak hastaların çoğunluğunda başlangıç tedavisine yanıt alımr. Son yıllarda, standart dozlarda kemoterapi ile karşılaştınldığında, alkilleyici ajanlarla yüksek doz kemoterapi kullanımından sonra otolog periferik kök hücre infüzyonu uygulamasının sağkalımı ve yaşam kalitesini arttırdığı gösterilmiştir. Bu yaklaşım küratif olmamakla birlikte, bazı hastalar için önemli bir tedavi seçeneği sunar ve ileri yaş hastalarda kabul edilebilir toksizitesi olduğu gösterilmiştir. Allogenik kemik iliği transplantasyonu miyelomlu hastalarda tek potansiyel küratif tedaviyi temsil edebilir, ancak ileri yaşlı veya ağır bir şekilde ön tedavi alan hastalarda eşlik eden aşırı morbidite ve mortalite bu hastalıkta kullanımını kısıtlamıştır. Standart tedaviden veya transplantasyondan sonra nüks gösteren hastalar alternatif kemoterapi rejimieriyle veya bortezomib (proteasome inhibitörleri sınıfında yeni orijinal bir ilaçtır) ile tedavi edilebilirler. Bortezomib aşırı bir şekilde tedavi edilen miyleomlu hastaların üçte birinde, bazı tam yanıtlar da dahil, yanıt almayı teşvik eder. Bortezomib asteni, trombositopeni veya nöropatiye neden olabilir. Çok etkili ve daha az yan etkili talidomid analogları da geliştirilmiştir ve klinik çalışmalar yürütülmektedir. Yüksek doz kortikosteroidler veya deneysel terapiler de kemoterapi veya transplantasyona yanıt vermeyen hastalarda tedavi seçenekleridir. Myelomun öngörülen komplikasyonlarına yönelik destekleyici bakım bu hastalığın tedavisinde önemli bir yer tutar. Ağrıyı ve patolojik kırıkları azaltmak amacıyla kemik rezorpsiyonlarına karşı zoledronic asit veya pamjdronat gibi difosfonatların düzenli enjeksiyonları yapılabilir. Kemik lezyonları, özellikle yük çeken kemikleri tutanlar, ağrı kontrolu ve patolojik kırıkları önlemek için palyatif radyasyon uygulanabilir.
Vertebral kemik lezyonları spinal kord kompresyonlarına neden olabilir, sırt ağrısını ve nörolojik semptomları arttırabilir. Kord kompresyonu düşündüren herhangi bir semptom derhal spinal manyetik rezonans görüntülerne yapılmasını ve, eğer gerekli olursa, tutulan bölgeye lokal radyasyon uygulanmasını gerektirir. İntravenöz kontrast maddeler dahil, nefrotoksik ajanlardan kaçınılması böbrek yetmezliğinin önlenmesi açısından önemlidir. Hafif-zincir depolanmasından kaynaklanan akut böbrek yetmezliğinde protein yükünü hemen düşürmek için plasmaferez yapılması yarar sağlar. Bütün hastalara pnörriokok ve H. injluenzae aşıları yapılmalıdır ve derin hipogamaglobulinemisi olan hastalarda yineleyen enfeksiyonları önlemek için intravenöz gamma globulin yararlı olabilir. Eritropoietin kullanımı anemiyi hafifletebilir ve kan transfüzyonları için gereksinimi azaltabiiir. WM plazmasitoid lenfositlerin malign hastalığıdır. Bu hücreler yüksek miktarlarda IgM üretimi gösterirler. İleri yaş hastaları etkileyen kronik bir bozukluk olup (medyan yaş 64 yıldır), düşük-dereceli lenfoma ve miyelom özelliklerini paylaşır. Miyelomun aksine, WM’de lenfadenopati ve splenomegali birlikteliği görülür ve kemik iliği tutulumu değişmez bir şekilde mevcut olmasına karşın, litik lezyonlar ve hiperkalsemi belirgin bir şekilde enderdir. WM’nin asıl klinik görünümü genellikle IgM’nin fiziksel özelliklerine bağlı olarak ortaya çıkan hiperviskozite sendromudur. IgG’nin aksine, IgM intravasküler alanda bulunur ve IgM düzeyleri yükseldikçe, plazma viskozitesi yükselir. Hiperviskozite semptomunun sıkça rastlanan semptomları; burun kanaması, retinal hemorajiler, baş dönmesi, konfüzyon ve konjestif kalp yetmezliğidir. IgM proteinlerinin yaklaşık %lO’u kriyoglobulin özellikleri taşır ve bu hastalar akrosiyanoz, Raynoud fenomen i ve vasküler semptomlar şeklinde görülen kriyoglobulinemi veya soğuk aglutinin sendromu semtomları görülür. WM’1i bazı hastalar neoplastik sürecin ortaya çıkmasından önce görülebilen periferik nöropati geliştirebilirler. WM’ye yaklaşım ve tedavisi diğer düşük dereceli Bhücreli lenfomalarınkine benzerdir. Nükleosid analogları (2-CDA ve f1udarabine) veya bir alkilleyici ajanın tek başına veya prednizonla kombine olarak kullanımı, adenopati, splenomegali ve M-zirvelerinin kontrolünde etkindir ancak küratif değildir. Rituksimabın, WM’de aktivitesi olduğu da gösterilmiştir. Plazmaferez akut olarak serum IgM düzeylerini düşürmekte etkindir ve sıklıkla hiperviskozite tedavisinin başında kullanılmalıdır. Tam remisyonlar enderdir. Terapiye yanıt veren hastaların medyan sağkalımları 4 yıldır ve bazı hastaların on yıldan fazla sağ kalımları olur.
Ağır zincir hastalığı ender görülen lenfoplazmasitoid neoplazmdır. Bu hastalığın karakteristik özelliği veya 1. tipinde bozuk ağır zincirlerin üretilmesidir. Klinik görünüm üretilen ağır zincirin tipiyle değişir. Gamma ağır zincir hastalığında yapısal semptomlara lenfadenopati, damak tutulumuyla birlikte Waldeyer halkası tutulumu eşlik eder. Alfa ağır zincir hastalığında, aynı zamanda Akdeniz lenfoması olarak ta bilinir, ince barsağın lenfoid infiltrasyonla tutulumu ve bununla bağlantılı olarak ishal ve malabsorpsiyon karakteristiktir. Mü ağır zincir hastalığı KLL ile birliktedir. Primer amiloidoz sistemik bir hastalık olup, immünglobulin hafif zincirlerinin organ ve dokularda depolanması sonucu organ işlev bozukluklarına bağlı bir grup semptomlar görülür. Konjestif kalp yetmezliği, kanama bozukluğu, nefrotik sendrom ve periferik nöropati yaygın komplikasyonlardır. Primer amiloidozlu hastalar miyelom için kullanılan tedaviye zayıf yanıt verirler. Eğer hastalar özellikle kardiyomiyopati gibi belirgin son organ hasarı gelişiminden önce tedavi edilirlerse, yüksek doz kemoterapi ve otolog kök hücre desteği ile cesaretlendirici sonuçlar alınmıştır.

Lenfosit olgunlaşmasını ve işlevini bir dizi konjenital bozukluklar etkiler ve immün yetmezlik bozuklukları ile sonuçlanır. Edinsel lenfosit işlev bozuklukları doğmalık bozukluklarla karşılaştırıldığında çok daha yaygındır. HIV en önemli edinsel immün yetmezlik nedenidir. HIVenfeksiyonlu hastaların NHL geliştirme riski yüksektir. HIV üzerine gelişen NHL’lar yaygın agresif B-hücre histolojik yapısındadır (yaygın büyük B-hücreli lenfoma ve Burkitt lenfoma dahil); bunlar yine sıklıkla EBV ile de bağlantılıdır ve başlangıçta tanı konulduğunda genellikle ekstranodal alanların tutulumuyla birlikte ileri evrededirler (evre III veya IV). HIV bağlantılı NHL’lı hastalar genel nüfustaki NHL alt tiplerinin tedavisinde kullanılan çoklu ilaç kemoterapi rejimIeriyle potansiyelolarak tedavi edilebilirler. Ek olarak, altta yatan HIVenfeksiyonun yüksek antiretroviral aktiviteli ilaçlarla tedavisi HIV’in eşlik ettiği NHL’li hastaların prognozunu ve hastalığın sonucunu iyileştirmiştir. Allogenik organ transplantasyonu yapılan hastalarda, kemik iliği alıcılarında graft versus host reaksiyonu veya solid organ transplantasyonu yapıldığında allograft rejeksiyonu engellemek için, güçlü immün baskılayıcı ilaçların kullanımı gerekir (örn., siklosporin, tacrolimus, azatiopirin, kortikosteroidler, metotreksat). Bu ilaçlar beraberinde edinsel immün yetmezlik durumun eşlik ettiği T-hücre işlevinde büyük kusurlara yol açabilirler ve transplant alıcıları bir takım viral ve protozoal enfeksiyonlara yatkındırlar. Ek olarak, güçlü immün baskılayıcı ilaçlar alan hastalar lenfoproliferatif bozukluk geliştirme riski taşırlar (posttransplant Iymphoproliferative disorder, [PTLD], transplantasyon sonrası lenfoproliferatif bozukluk). PTLD kinik olarak agresif lenfoma gibi davranır. PTLD, monoklonal veya poliklonal olabilen B hücrelerin polimorf veya monomorf topluluklarıyla karakterize olan EBV ilişkili lenfoproliferatif bozukluktur. PTLD geliştiren hastalar mümkün olduğunda immün baskılayıcı tedavinin dozu düş.ürülerek tedavi edilirler; yalnız başına bu müdahale ile sitotoksik terapiye gereksinim önlenerek, hastaların yaklaşık yarısında PTLD’nin gerilemesi sağlanır.
Lenfositler enfeksiyona karşı konak yanıtında gerekli rol oynar. Bu yanıt klinik olarak periferik kandaki lenfositlerde artış (reaktif lenfositoz) ve lenf düğümü büyümesi şeklinde görülebilir. Reaktif lenfositoz daima poliklonaldir, genellikle T-hücre hakimiyeti olur, genellikle yaygın monoklonal B-hücreli neoplastik süreçlerden kolayca ayırt edilebilider. Bazı enfeksiyonlara tipik olarak belirgin lenfositoz eşlik eder (örn., EBV’nin eşlik ettiği enfeksiyöz mononükleosis, sitomegalovirüs, immün baskılanmış konaklarda toksoplasmozis, viral hepatit). İster bölgesel ister yaygın lenfadenopati şeklinde olsun, lenf düğümlerinin büyümesi bazı enfeksiyonların ortak belirtisidir. Lenfadenopati ve hassasiyet eşlik eder. Birçok olguda, adenopati reaktiftir ve kültürde üreme genellikle olmaz; diğer durumlarda (örn., tuberküloz veya fungal enfeksiyonlar) lenf düğümü dokusunda kültür veya uygun boyama organizmayı aydınlatabilir. Düğümden alınacak biyopsi genellikle sürecin neoplastik olmayan doğasını doğrulayacaktır; normal yapı ve hücresel dağılım ve lenfüid hücrelerin monoklonal yapıda olmayışı biyopsi özellikleridir.