
Malign dönüşümler, hücresel olgunlaşma ile farklılaşma bozukluklarının kombinasyonunu içermektedir. Onkojenezin çoklu adım teorisi, bu eksikliklerin birbirinden ayrılabildiğini ve normalden tam olarak dönüşüme uğramış hücreye kadar tüm aşamalardaki bozuklukları kapsadığını düşündürmektedir. Hematopoetik hücrelerin sürekli yenileniyor olması, bu modeli destekleyen klonal genetik anomalilerin gelişebilmesi için uygun ortam sağlamaktadır. Hematopoetik kök hücresinin klonal bozuklukları bir dizi prelösemik ve lösemik duruma yol açmaktadır. Birincilolgunlaşma bozuklukları, myelodisplastik düzensizliğe neden olurken, proliferasyonun normal kontrolünün kaybolması myeloprolifemtif ·hastalık ile sonlanmaktadır. Tüm bu bozukluklar prelösemik olup, akut lösemi ye dönüşmeleri kesin, ancak dönüşme hızları değişkendir.
MYELODİSPLASTİK SENDROM
Etyolojisi ve patogenezini incelediğimiz zaman myelodisplastik sendromlar (MDS), miyeloid hücre serisinin bir veya daha fazla kolunda yetersiz ve bozuk hematopoezis ile karakterize heterojen kan hastalığıdır. Kemik iliğinde hematopoetik hücre sayısı normal veya artmış olmasına rağmen hastalarda pansitopeni görülür. Olgunlaşma bozukluklarına, olgun hücrelerin perifere salınımmın azalmasına katkıda bulunan intramedüller apopitoz (programlanmış hücre ölümü) artışı eşlik etmektedir.
Birincil MDS ağırlıklı olarak yaşlı kişilerin hastalığıdır ve 60-75 yaş arası hastaların yaklaşık 500’de birinde görülmektedir. Çoğu vaka idyopatiktir ve tüm vakaların yaklaşık olarak %25’i akut myelojen lösemi veya myeloid lösemi ye dönüşme riski taşımaktadır. Daha önce radyasyona, kemoterapiye ve organik kimyasallara (benzen) maruz kalmış kişilerde, MDS insidansı artmaktadır.
İkincil MDS, kemoterapi (alkilleyici ajanlar, antrasiklinier), iyonize radyasyon ve kemik iliği transplantasyonu sonrası her yaşta hastada gözlenebilir ve tanı konmuş tüm MDS vakalarının %10 ile 20’sini kapsar. Terapi sonrası oluşan MDS mjyelotoksik terapiden en az 6 ay sonra ortaya çıkar. Sitogenetik anomalilerin eşlik ettiği kötü bir prognozu vardır ve hızla AML’ye dönüşür. Primer kanser tedavileri yaşam süresini uzatırken, ikincil myelodisplazi insidansını da artırmaktadırlar.
Tanısını koyarken MDS’li hastaların çoğu, tesadüfi olarak saptanan periferik sitopeninin değerlendirilmesi için sevk edilirler. Semptomatik hastalarda bulgular, genellikle, sitopeninin ikincil etkilerine bağlı olarak gelişmektedir; trombositopeniye bağlı kanama ve çürükler, lökopeniye bağlı enfeksiyon ve anemiyle ilintili olarak yorgunluk, halsizlik ve dispne bulunmaktadır. Fizik muayenede, belirgin bir şey bulunamamakla birlikte, hastaların %25’i veya daha fazlasında splenomegali saptanmıştır. Bazı hastalarda cilt lezyonları ile birlikte görülen ateş (akut febril nötrofilik dermatoz veya Sweet sendromu) MDS’nin akut lösemiye dönüşmesinin habercisi olabilmektedir.

Sitopenilere ek olarak, periferik yaymada karakteristik morfolojik anormallikler bulunmaktadır. Eritroid hücreler, bazofilik noktalı ve makrositiktir. Nötrofiller ise sıklıkla hipogranüler ve az lobludur. Psödo Pelger Huet anomalisi olarak bilinen iki loblu çekirdek karakteristiktir. Periferik saymada çok sayıda bant tespit edildiğinde Pelger Huet anomalisinden şüphelenilmelidir.
MDS’de kemik iliği genellikle normoselüler veya hiperselülerdir, ancak hastaların % 1O’unda kemik iliği hiposelüler olabilir. Displastik değişiklikler her üç hücre grubunda da gözlenebilir. Eritroid hücreler, çok çekirdekli veya senkronu kaybolmuş çekirdek-sitoplazma gelişimi olan megablastlar haline gelmişlerdir. Anormal küçük mikromegakaryositler ve agranüler megakaryositler bulunmaktadır. Myeloid seride erken myeloid formlara doğru sola kayma saptanır. Myeloblast sayısı sıklıkla artmıştır ve bu hücreler akut lösemiye doğru ilerlemenin göstergesidir.

Elektron mikroskobu ile bakıldığında kemik liğinde artmış apopitozun karakteristiği olan hücresel değişiklikler (belirgin çekirdek kromatini, sitoplazmik vakuoller ve baloncuklar) saptanmaktadır. MOS tanısı erişkinlerde kemik iliği displazisine neden olan pek çok nedenden ayrılmalıdır. Klonal sitogenetik anomaliler tespit edilememişse, akut hastalık durumlarında, uzun süreli hastane yatışlarında veya 6 aydan daha uzun süreli radyasyon ve kemoterapi gibi miyelotoksik tedavi görenlerde MOS tanısı asla konmamalıdır.
Vitamin B12 ve folat eksikliği, alkol kullanımı, kemoterapi ve insan immün yetersizlik virüsü enfeksiyonu gibi kemik iliği displazisi yapan nedenler de dikkate alınmalıdır. Kemik iliği hiposelüler olan ve MOS 'den şüphelenilen bir hastada, sendromun aplastik anemiden ayrılması gerekmektedir. Kemik iliğinin sitogenetik analizi, tüm hastaların üçte biriyle yarısında klonal kromozomal anormallikler göstermektedir ki, bu hastalarda MOS için tanı koydurucudur. MOS’de karakteristik olarak gen delesyonlarının (5q-) ve AML alt tiplerinde karakteristik olarak translokasyonların saptanmış olması, klonal myeloid kök hücre hasarlarında da benzer mekanizmaların rol oynadığını düşündürmektedir.
Geçmişte MOS, Fransız-Amerikan-İngiliz (FAB) sistemine göre, ilik hücre morfolojisine ve blast oranına dayanılarak beş alt tipe sınıflandırılmaktaydı: dirençli anemi(RA), halka şeklinde sideroblastlarla giden dirençli anemi (RARS), aşırı blastlarla giden dirençli anemi (RAEB), dönüşüm halinde blastlarla giden dirençli anemi (RAEB-T) ve kronik myelomonositik lösemi (CMML). AML de %30’dan fazla blast varlığı ile tanımlanmaktaydı. Yakın zamanda Dünya Sağlık Örgütü (OSÖ) FAB kriterlerine yeni kemik iliği ve genetik bulguları ekleyerek yeni bir sınıflama kriterleri yayınlamıştır. Blast sayısı %20’yi geçen hastalar ile %30’u geçen hastaların durumları aynı gözlemlendiği için OSÖ, AML’nin tanısını kanda veya kemik iliğinde %20 den fazla blast görülmesi olarak yeniden tanımlamıştır.
FAB sınıflamasına göre 5 alt gruba ayrılan MOS multi kökenli displazinin tanımlaması ile (örn., refrakter sitopeni ile birlikte ımılti kökenli displazi, refrakter sitopeni ile birlikte multi kökenli displazi ve sideroblastlar) 8 alt gruba ayrılmış ve CMML myeloproliferatif-myelodisplastik sendrom olarak sınıflandırılmıştır. Myelodisplastik kemik iliğinde izole 5q- sitogenetik anomalisinin varlığı; anemi, normal veya artmış trombosit sayısı ve yavaş ilerleyen lösemi ile karakterize ayrı bir klinik sendrom olarak kabul edilmektedir. Daha da önemlisi AML’nin belirli alt sınıfları kemik iliği blast sayısından bağımsız olarak yeniden tanımlanmıştır.
Osplastik kemik iliğindeki klasik karyotipik anormalliklerin tanımlanması, özellikle t(8;21), inv(16) ve t(15;17), AML tanısının konması için artık yeterlidir. Bundan başka kemoterapi, radyasyon veya diğer miyeloablazif tedavilerin sonrasında ortaya çıkan ilik displazisi çabucak akut lösemiye dönüştüğünden, MOS’den çok tedavi-ilişkili AML (t-AML) olarak değerlendirilmektedir.
Prognoz ve tedavisini incelediğimiz zaman MOS’nin klinik seyri değişkendir. Bazı hastalar normal yaşamlarını sürdürürken, çoğu sitopeniyle ve/veya kemik iliği yetmezliğine bağlı komplikasyonlar nedeniyle daha erken ölürler. MOS’li hastaların % 15-20’si AML’den ölmektedir. AML’de (ileriye bakınız) olduğu gibi, bazı MOS alt tiplerinin doğal seyri ve tedavisi belli bazı sitogenetik anomalilerle ilişkilidir ve bu nedenle kemik iIiğinin moleküler incelemesi başlangıç değerlendirmesi olarak yapılmalıdır. Örneğin, 5. kormozomun uzun kolunda izole delesyonu olan hastalarda (5q- sendromu) klinik seyir belirlidir. Bu hastalar, genellikle dirençli makrositik anemisi, normal veya artmış trombosit sayısı olan, daha iyi klinik seyre sahip yaşlıca kadınlardır. Bu hastalar aralıklı verilen kırmızı kan hücre transfüzyonu ile uzun yıllar yaşarlar ve hastalığın lösemi ye dönüşme riski düşüktür.
Tersine; MOS’ye kromozom Tnin kısa kolunda delesyonun (7p-) veya monozomi 7 veya trizomi 8 gibi kompleks anomalilerin eşlik etmesi kötü prognoz belirtisidir. Genellikle dirençli anemisi ve fazlaca bl astı veya çoklu köken displazili dirençli sitopenisi olan hastaların şansı düşüktür. Ancak bu sınıflama aşağı yukarı sadece yaşam süresi ile korelasyon göstermektedir.
1998’de Uluslararası MOS Risk Analiz Çalışma Grubu, MOS’nin prognostik ölçütlerini daha iyi belirlemek amacıyla Uluslararası Prognostik Skorlama Sistemi’ni (IPSS) geliştirmiştir. lPSS, MOS hastalarını, sitogenetik anormali, sitopeni, ilerlemiş yaş ve kemik iliğinde blast oranı gibi çeşitli değişkenlere dayanarak 3 pragnostik kategoriye ayırmaktadır. Bu kategoriler lösemiye dönüşüm süresi ve yaşam beklentisi ile ilintilidir.

Malesef; MOS tedavisindeki seçenekler sınırlıdır ve büyük oranda hastanın yaşı, performans durumu, hayat kalitesi, hastalığının şiddeti ile IPSS ve sitogenetik tarafından tanımlanmış pragnostik faktörler tarafından belirlenmektedir. MOS hastalarının büyük çoğunluğunu oluşturan yaşlı hastalar, iyileşme umudu olmaksızın, saldırgan girişimleri tolere edemeyebilir veya istemeyebilirler. Hastalığın dönüşme riskinin düşük bulunduğu hastalarda, en iyi tedavi, eritrosit ve trombosit transfüzyonudur. Kronik transfüzyonlar ise demir yüklemesine yol açar çünkü her ünite eritrosit ile hastaya 200-250 mg demir verilmiş olur. Fazla demir makrafajlarda depolanır ve sonunda karaciğer parankimi, miyokard, deri ve pankreasta birikerek sekonder hemokromatozise yol açar. Sonuçta karaciğer yetmezliği, kalp yetmezliği, hiperpigmentasyon ve diabetes mellitus gelişebilir.
Komplikasyonları önlemek için transferin satürasyonu ve serum ferritin seviyeleri belirgin olarak yükselen hastalara deferoksamin (hergün birkaç saat subkutan veya intravenöz infüzyon şeklinde,) veya yeni oral şelat Eksjade tedavisi uygulamak gerekir. Bazı hastalarda rekombinan büyüme faktörleri (granülosit-koloni uyarıcı faktör, granülosit-monosit koloni uyarıcı faktör, eritropoetin [EPO], gibi) tek başlarına veya kombine olarak uygulandıklarında transfüzyon ihtiyacı azalmaktadır. Özellikle endojen serum EPO seviyesi düşük olan hastalar çok fayda görürler. Bu tedaviler palyatiftir ve beklenen yaşam süresini etkilemezler.
Yüksek riskli MOS hastaları (lösemiye öncüllük eden sitogenetik anormalileri ve/veya dolaşım kanında blast sayıları fazla olan hastalar) saldırgan AML-temelli kemoterapiye adaydırlar. Ancak, MOS veya MOS-ilişkili AML’nin standart kemoterapi rejimIeriyle iyileşme hızı düşük olmakta; hastalıksız dönemler kısa sürmekte, ilk i2- i8 ayda nüks hızı artmakta ve tedaviyle yaşam süresini uzatmak, iyileşen hastalarda bile olanaklı olmamaktadır.

Diğer hematolojik kök hücre bozukluklarında olduğu gibi, tek iyileştirici tedavi allojenik kök hücre transplantasyonudur. Bu tedavi idealolarak tamamen iyileşmiş hastalara uygulanmalıdır. 40 yaş altındaki ve insan lökosit antijeni (HLA) uyumlu kardeş vericileri olan tüm hastalara bu işlem önerilmelidir. Düşük hastalık riski olan bu tip hastalarda uzun dönem hastalıksiz yaşam süresi %50’lerin üzerindedir. İndüksiyon kemoterapisi veya kök hücre nakli için uygun olmayan veya istemeyen MOS’li hastalar hastalığın biyolojik mekanizmalarını hedef alan tedavilerden fayda görürler. Düşük risk MOS’si veya spesifik HLA (HLA-OR 15) haplotipi olan genç hastalar, antİtimosit globülin veya siklosporin-A gibi T hücrelerini baskılayan tedavilerinden fayda görürler ve kan sayımında %30-50 iyileşme gözlenir. Bu durum kemik iliğindeki baskılanmanın otoimmün olabileceğini düşündürmektedir.
Talidomid veya türevi olan lenalidomid gibi daha yeni bağışıklık düzenleyici ajanlar MDS hücrelerinin ve ilik mikro ortamının büyümesini engellerler ve hastaların üçte bir kadarında olumlu cevap yaratırlar. 5q-sendromu olan hastalar özellikle lenalidomid tedavisinden fayda görürler ve tedavi sonrası %66’lara varan tam ve uzun süren bir cevap gözlenir; ilikte anormal sitogenetik hücre klonu kaybolur. Daha yaşlı hastalarda decitabine veya 5-azasitidin gibi DNA metiltransferaz inhibitörleri anormal hipermeti lasyonunu geri çevirebilir ve lösemi dönüşümüne katkıda bulunan geni susturabilir. Bir çalışmada yaşlı, 5-azasitidin uygulaması nakile bağlı MDS’si olan hastaların üçte ikisinde lösemiye dönüşümü belirgin olarak geciktirmiş ve sadece destek tedavisi olan hastalara kıyasla yaşam kalitesini kayda değer biçimde artırmıştır. Tedavi etmese de, bunlar gibi ümit veren biyolojik tedaviler MDS’nin doğal seyrini etkileyerek yaşam süresini uzatabilir.
Kronik Miyeloproliferatif Bozukluklar
Kronik myeloproliferatif bozukluklar (MPD); lökositoz, trombositoz, eritrositoz, splenomegali ve kemik iliği hiperselüleritesi ile karakterize klonal kök hücre bozukluklarıdır. Hematopoetik hücre kitlesini düzenleyen normal geri bildirim mekanizmalarının uyarılmasına neden olan multipotent kök hücrenin bozukluğu MPD için önemli bir noktadır. MPD’li hastalardan alınan kök hücreler in vitro ortamda ekzojen sitokinler olmaksızın serum ile in vitro koloni büyümesi göstermişlerdir ve bu teknik MPD için tanı testi olarak kullanılmaktadır. MPD için polisitemi vera (PV), esansiyel trombositoz (ET), kronik idiyopatik miyelofibrozis (IMF, agnojenik myeloid metaplazi olarak da bilinir) kronik myelojen lösemi (KML) şekilinde hiperproliferatif hücre tipine göre bir sınıflandırma yapılmıştır. Hipereozinofilik sendrom (HES) ve KML de yakın zamanda myeloproliferatif bozukluklara eklenmiştir
MPD’ye bağlı komplikasyonlar bir veya daha fazla hücre neslinin aşırı çoğalmasından kaynaklanmakta ve hepsine, KML hariç, geç ve nadir gelişen bir komplikasyon olan akut lösemi eşlik edebilmektedir. MPD’nin patogenezi hastaların çoğunda disfonksiyonel kinazlara dayandırılmaktadır. KML’de Philadelphia kromozom anomalisi, yapısal kinaz aktivitesi olan breakpoint cluster region-abelson lösemisi (ber-abl) füzyon proteini oluşumuna yol açarken; PV, IMF ve ET’de hastaların çoğunda Janus kinaz 2 (JAK2)'nin 617. pozisyonunda (V617) fenilalanin ile valinin yer değiştirmesine yol açan bir mutasyon saptanmıştır ve kök hücre bozukluklarının anormal büyüme özelliklerine bu durum neden oluyor olabilir.
POLİSİTEMİA VERA
PV, genel anlamıyla kanda eritrositlerin artmasıdır. Klonal multipotent kök hücre defektinden kaynaklanan periferde artmış eritrosit kitlesi ile karakterize bir sendromdur.Hastalarda birim hacim başına hemoglobin yüksekliği (eritrositaz) ilk teşhis edildiğinde yapılacak ilk değerlendirme bunun gerçek bir eritrosit artışı mı (mutlak eritrositoz veya polisitemi) yoksa azalmış plazma hacmi ile beraber normal eritrosit sayısı mı (dehidrasyon veya başka sebeplere bağlı nisbi eritrositoz) olduğunu bulmak olmalıdır. İkinci durum gerçek polisitemi değildir. Polisitemi veya mutlak eritrositoz artmış eritrosit yapuruna bağlı olarak eritrosit kitlesinde mutlak bir artışı ifade eder. Vücudun eritrosit üretebilme yeteneği hipokserru, anerru ve akut kan kaybı durumlarında, dokulara sürekli oksijen iletilebilmesim garantilemektedir.
Fizyolojik uyarılara cevaben pluripotent kök hücre öncülleri EPO tarafından eritroid progenitor hücrelere ve sonunda da hemoglobin taşıyan olgun eritositlere farklılaşmak üzere uyarılıdar. Yeterince kırmızı küre üretildiğinde; negatif geribildirim mekanizması EPO üretirruni baskılar ve serum hemoglobin seviyesi normalolarak kalır. Normal erken eritroid progenüor hücreler, böbreğin hipoksiye cevaben ürettiği bir büyüme filiörü olan, pluripoten kök hücrelerinden farklılaşmak üzere uyanlmaktadırlar. Polisiterru, periferik kanda eritrosit kitlesimn artması olarak tanımlanmaktadır. Kök hücre bozukluğuna bağlı birincil veya fizyolojik uyaranlara cevaben eritropoeziste artmaya bağlı ooncil olabilmektedir.
PV sebebi bilinmeyen, diğer hematopoetik anormalliklerin de bulunabildiği ancak eritrositozun başrolde olduğu bir klonal kök hücre hastalığıdır. Tüm hastalann yansında eş zamanlı lökositoz ve/veya trombositoz görülmektedir. Ancak eritrositoz bu hastalığın karakteristik özelliğidir ve pek çok ciddi klinik komplikasyonun da sebebidir. PV tamsı daha önce, artmış eritrosit kitlesi, splenomegali, trombositoz, lökositoz, hipokseminin ve polisiteminin ooncil nedenlerinin yokluğu, artmış lökosit alkalin fosfataz ve serum vitamin BI2 seviyelerine dayanan bir dışlama tanısıydı. Son zamanlardaki hastalığın patogenezi ile ilgili bilgiler tanı kriterlerinde değişikliğe yol açmıştır.
Anormal bir kemik iliği klonunun saptanması, düşük serum EPO seviyesi ve in vitro eritroid kolonilerinin EPO’dan bağımsız olarak koloni büyümesimn gerçekleşmesi günümüzde primer PV tanısının konulmasına çok yardımcıdır. Son zamanlardaki yayınlarda PV’li hastaların çoğunluğunda EPO reseptörüne bağlı JAK2’de aktive olmuş mutasyonlann varlığı gösterilıniştir. Klinik ve Laboratuvar Çalışmaları PV 100.000 kişiden 1 ila 3’ünde görülür ve ortalama başlangıç yaşı 65’tir. PV’mn erken tanısı ve tedavisi önemlidir çünkü tedavi edilmeyen hastalarda serebral, koroner ve mezenterik dolaşırnda tromboembolik hastalığa bağlı ciddi morbidite ve mortalite riski çok artmaktadır. Hastalann %20’sİ arteryel ve· venöz trombozia başvurmaktadır ve trombozis en sık ölüm sebebidir.
Tipik olarak hastalar, baş ağrısı, görme sorunlan, mental bulutlanma ve banyo sonrası kaşıntıdan yakınmiliadular. İnme, geçici iskemik ataklar, miyokard iskemisi gibi tıkayıcı vasküler olaylar ve parmak ağrısı, paresteziler veya gangren yaygındır. Ek olarak; pulmoner, derin ven, hepatik ve portal ven trombozları görülebilir. Paradoksik olarak; hastalar trombosit işlevlerinde bozulmaya bağlı olduğu samlan hemorajik olaylara yatkın hale gelirler ve böyle hastalar gastrointestinal kanamayla başvurabilirler. Fizik muayenede, sıklıkla retİnal ven tıkanıklığı, splenomegali ve kırnuzı siyanoz saptanır. Periferik kan, demir eksikliği olsun veya olmasın, mikrositiktir. Kemik iliği hiperselülerdir. Tanı sırasında, sitogenetik özellikler sıklıkla normaldir; klonal sitogenetik anormalliklerin gelişmesi hastalığın ileri aşamalarına geçeceğinin habercisidir. JAK2 (V617) mutasyon insidansının PV’de %65 ile %97 arasında değiştiğini gösteren çeşitli çalışmalar vardır.
Tedavi olmazsa, PV’lı hastaların yarısı tanı konulduktan sonraki 18 ay içinde trombotik komplikasyonlar nedeniyle ölmektedirler. Tedaviyle bile PV, kronik, ilerleyici bir hastalıktır. Miyelofibroza ve miyeloid lösemiye dönüşme riski, 20 yıl içinde,%5-20’dir. İleri yaşta, tromboz öyküsü olan ve hematokrit değeri yüksek olan hastalarda vasküler olay riski yüksektir. Bu nedenle de; aralıklı flebotomi tedavinin temel taşıdır.
Flebotomi demir eksikliği anemisine neden olarak eritrosit yapım hızını daha da düşürür. Semptomatik splenomagali veya tromboz riski taşıyan ve flebotominin başarısız olduğu hastalarda sitoredüktif terapi endikedir. Geçmişte lökositoz ve trombositozun tedavisi için kullanılan düşük doz kemoterapötik ajanların (klorambusil, busulfan gibi) veya radyoaktif fosfor (32P)'un toksisitesi yüksekti ve sekonder AML riskini artırıyordu. Günümüzde, ağırlıklı olarak hidroksiüre (lösemi riskini artırmadığı düşünülen düşük doz sitotoksik bir ajan), interferon-a tedavisi (genç hastalarda veya hamile kadınlarda tercih edilir) ve anagralit (dirençli trombositoz tedavisi için kullanılan megakaryotoksik ajandır) kullanılmaktadır.
Terapinin amacı; hematokriti kadınlarda %42’nin, erkeklerde %45’in altında tutmaktır. Tüm myeloproliferatif hastalıklarda olduğu gibi sitoredüktif terapi ye başlanması hiperürisemiye neden olabilir (sekonder gut ve ürik asit taşlarına yol açar); bu da allopürinol ile tedavi edilebilir. Düşük doz aspirin ve asempetomatik trombositozun tedavisi PV’li hastalarda tromboembolik olayların azalmasını sağlar ve özellikle kardiyak risk taşıyan yaşlı hastalarda önemsenmelidir. Daha genç hastalarda steroid olmayan anti-inflamatuar ve anti-trombotik ilaçlar, gastrointestinal kanama riski nedeniyle dikkatli kullanılmalıdırlar. Etkili tedaviyle bu hastalarda uzun dönem yaşam süresi çok yüksektir.
Esansiyel Trombositoz
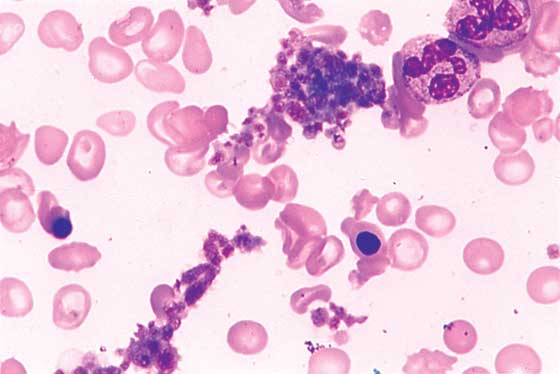
Esansiyel trombosİtoz (ET), trombosit ve lökosit sayısında artma ile sonuçlanan pluripotent kök hücre bozukluğudur. Trombosit işlevi ve yaşam süreleri normaldir. Trombosit sayısındaki artış altta yatan sebebe ikincil (bakteriyel enfeksiyon, sepsis, demir eksikliği, otoimmün hastalıklar ve diğer malign hastalıklar) olabileceğinden, ET tanısı konulmadan önce hastalığa neden olabilecek diğer hastalıklar dışlanmalıdır. Genelde, tanı 600.000x ıo9/1t üzerinde trombosit sayısı, normal eritrosit kitlesi, normal demir düzeyi ve kemik iliği incelemesinde megakaryositik seriye ait proliferasyon, çok sayıda olgun megakaryosit ve az sayıda, eğer varsa, displastik veya mikromegakaryosit ile konur.
Kemik iliğinde immünohistokimyasal ve sitogenetik çalışmalar ile myelodisplazi, myelofibrozis ve KML için tanısal değeri olan Philadelphia kromozomu ekarte edilmelidir. ET için %100 spesifik genetik veya biyolojik marker tanımlanmamış olsa da JAK2 V617 mutasyonunun hastaların yarıdan fazlasında gösterilmiş olması, ET yi V617F pozitif veya V617F negatif olarak alt tiplere sınıflandırılması önerilmektedir. Diğer MPD’lerden farklı olarak, ETli hastaların kemik iliğinde, sıklıkla faktörden bağımsız koloni büyümesine rastlanmamaktadır ve bu hastalığın kesin sebebi (ve JAK2 mutasyonları ile olan ilişkisi) yoğun bir biçimde araştırılmaktadır.
ET, vaka sayısının asemptomatik hastalarda rutin laboratuar incelemesi sırasında tespit edilmesiyle arttığı, nadir görülen bir bozukluktur. Hastalığın ortalama ortaya çıkış yaşı 60-65 olmakla birlikte, hastaların % 10-25’i 40 yaşın altındadır. Hastaların üçte iki kadarı semptomatiktir. Vazomotor semptomlar; baş ağrısı, sersemlik, görme değişiklikleri ve eritromelajiyi (el ve ayaklarda yakıcı ağrı ve eritem) içermektedir. Geçici iskemik ataklar, inme, nöbet, anjina ve miyokard enfarktları gibi ciddi arteryel trombotik komplikasyonlar görülebilir. Nadiren hastalarda kaşıntılı deri lezyonları ve hematomlar gelişir. Gastrointestinal kanama riski %5’in altındadır. Genelde, bu bozukluğu olan hastaların uzun dönem hayatta kalma hızları, yaş açısından eşlenmiş kontrollere benzer seyretmektedir. Diğer MPD’lere göre lösemiye dönüşme riski oldukça düşüktür (%3-4). Ancak, tekrarlayan hemorajik ve trombotik komplikasyonlara bağlı morbidite yüksektir ve trombosit sayısına veya işlevlerine bakarak tahmin etmek mümkün değildir. Tedavi ömür boyu devam edeceğinden, risk faktörleri ile önceki klinik bulgu ve belirtiler tedaviyi yönlendirir.
Kardiyovasküler risk faktörlerinin (sigara, hipertansiyon, şişmanlık ve (hiperkolesterolemi) agresif tedavisinden bütün hastalar yarar görür. Düşük doz enterik aspirin nörolojik semptomların rahatlatılmasında etkili olup, kanamaya neden olma riski düşüktür. Genç ve/veya hamile hastalar semptomatik hale gelene kadar tedavi edilmedikleri halde, daha yaşlı (>60 yaş), tromboz öyküsü olan veya hastalığı uzun süredir devam hastalar trombosit sayısını düşüren tedavilerden fayda görürler.
Özgül olmayan miyelosupresif bir ajan olan hidroksiüre genellikle ilk tercih edilen ilaçtır ve uzun dönem lökomojenik riski iyi tolere edilir. Anagrelit (trombosit aggregasyonunu ve mega karyosit olgunlaşmasını inhibe eden oral ajan) eğer hidroksiüre akut yan etkiler (sıvı retansiyonu, palpitasyonlar), kanama (beraberinde aspirin kullanılmışsa) ve miyelofibrotik dönüşüm riski gibi nedenlerle başarısız olursa kullanılacak ikinci tercih ilaçtır. Her iki ilaç da teratojeniktir ve ET’li hastaların önemli bir bölümünü oluşturan çocuk doğuracak yaştaki genç kadınlarda kullanılmamalıdır. ET’li hastalarda fetus kaybı yüksektir. İnterferon-a (malign klonun biyolojik mekanizmalarına etki eden ancak plasentayı geçmeyen bir sitokin) heparin ve aspirin ile kullanıldığında haıruleliğin başarılı biçimde sonuçlanma şansını arttırmaktadır.
KRONiK İDİYOPATİK MİYELOFİBROZ
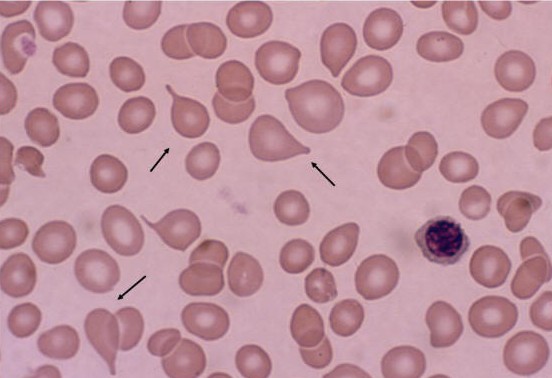
Kronik IMF (agnojenik miyeloid metaplazi olarak da bilinir), aşırı düzeyde ilik fibrozu ve sonucunda gelişen ilik yetmezliğiyle karakterize klonal kök hücre bozukluğudur. Anormal bir myeloid öncülün; artmış miktarlarda fibroblast büyüme faktörü üreten displastik megakaryositlere öncülük ettiğine inanılmaktadır. Bu sitokinler normal fibroblast ve diğer stroma hücreleri üzerine etki ederek, aşırı proliferayonu ve kollajen depolanmasını uyarmaktadırlar. Zamanla, kemik iliğinde fibrozisin artması, multipotent hematopoetik öncüllerin perifere erken salınmasına yol açmaktadır. Bu hücreler, daha sonra diğer alanlara göç eder ve kendilerini tekrardan oluştururlar, böylece hematopoez kemik iliğinden diğer dokulara, özellikle de karaciğere ve dalağa kayar. Bu süreç, ekstramedüller hematopoez olarak adlandırılır.
Hastalığın erken evrelerinde hastalarda anormal kan sayımları rutin laboratuvar testlerinde görülebilir. Kompansatuar ekstramedüller hematopoez sonucu trombosit ve eritrosit hücre sayıları artmış veya normal sınırlarda olabilir. Periferal kanda göz yaşı damlası şeklinde eritı-ositler, dev trombositler ve immatür nonlösemik miyeloid, eritroid ve lökosit hücreleri gibi lökoeritroblastik değişikleri görülebilir. İMF tanısı normal eritrosit kitlesi içeren kemik iliği fibrozisi, Philadelphia kromozom yokluğu (KML için tam koydurucu), splenomegali, anemi, ekstramedüller hematopoeze ait delillerin varlığıyla konulabilir. Kemik iliği fibrozuna neden olan hem neoplastik hem de non-neoplastik nedenler elenmelidir.
Miyelofibroz (agnojenik miyeloid metaplazi) yaşlı kişilerde rastlanılan nadir ve honik bir hastalıktır; yıllık insidansı 100.000’de 0.5’tir. Hastalığın başlangıcında, hastalar asemptomatik olabilir. Daha sonra, hastalar aneıniye bağlı ilerleyici yorgunluk ve dispneden; veya splenomegali ve üst kadran ağrısı/dolgunluğundan yakınırlar. Bu hastaların yarısından fazlasında heptamosplenoınegali gelişir. Hastalığı daha ileri dönemlerinde, hastalarda ateş, kilo kaybı ve gece terlemesi gibi yapısal semptomlar da gelişebilir.
Kemik iliği yetmezliği ilerledikçe, nötropeni ve trombositopeni komplikasyonları da gelişmektedir. Gizli dissemine intravasküler koagulasyona bağlı kanamalar da bir risk oluşturmaktadır. Peritonel ve plevral boşluklarda ve merkezi sinir sisteminde (MSS) ve omurilikte gelişen ekstramedüller hematopoez de semptomlara neden olabilir. Ortalama yaşam süresi kısadır, 2 ile 5 yıl arasında değişmektedir. Prognozu olumsuz yönde etkileyen faktörler lökosit sayısının 4000’nin altında veya 30.000’nin üzerinde olması, dolaşımda blast yüzdesinin yüksek olması, sistemik semptomların olmasıdır. Diğer önemli klinik faktörler 60 yaşın üzerinde olmak, trombositopeni, masif hepatosplenomegali ve sitogenetik anomalilerdir. Zaman içinde hastalık kronik fazdan daha hızlı bir faza geçebilir ve hastaların %8-10’unda lösemiye dönüşüm görülebilir. IMF sonrası gelişen AML’nin tedavisi genellikle yoktur.
Diğer lösemi harici ölüm nedenleri, kalp yetmezliği, enfeksiyon, intrakraniyal kanama ve pulmoner embolidir. Miyelofibroz için iyileştirici bir tedavi bulunmamaktadır. Genç hastalarda, deneysel aşamadaki HLA uyumlu kardeşlerden alınacak allojenik kök hücre transplantasyonu düşünülebilir. Semptomatik anemisi olan her hasta palyatif transfüzyon ve rekombinan eritropoietin, androjen (danazol) veya düşük doz talidomid uygulamasından fayda görür ve eritrosit düzeyini koruyabilmek için yapılmalıdır. Belirgin trombositoz ve lökositoza veya ekstramedüller hematopoeze bağlı semptomları olan hastalarda hidroksiüre (ilk tercih), interferon-a (daha genç hastalarda) veya busulfan, interferon-a, mefalan gibi düşük doz kemoterapötikler kullanılabilir. Semptomatik splenomegalisi, dirençli trombositopenisi, hipermetabolik semptomları ve portal hipertansiyonu olan kişilere splenektomi önerilmektedir ancak ameliyata bağlı morbidite ve mortalite oldukça yüksektir.
Cerrahi uygulanamayan hastalar, palliyatif dalak radyasyonundan veya anormalolan ilik mikroçevresini hedef alan geliştirilme aşamasındaki yeni biyolojik ajanlardan yarar görebilirler. Ancak, hiçbir tedavi yaşam süresini uzatmaz veya hastalığın ilerlemesini ciddi biçimde durdurmaz.
KRONİK MYELOİD LÖSEMİ
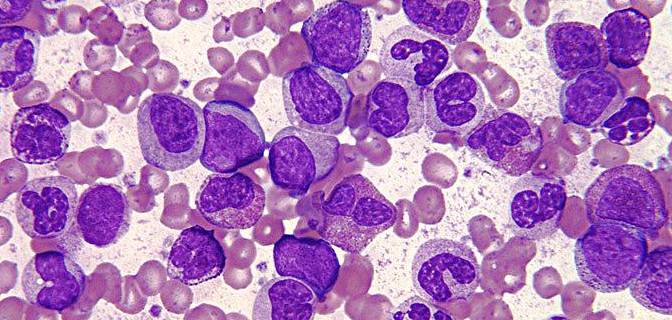
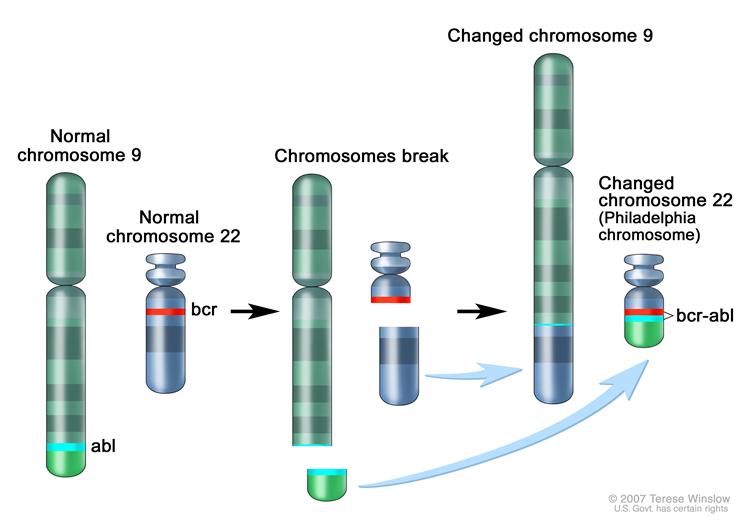
KML, artmış granülosit hücre serisi ve beraberinde eritroid ve trombositik hiperplazi ile karakterize bir MPD’dir. MPD’ler içinde, doğal öyküsü, önlenemez biçimde akut lösemiye dönüşüyor olması ile benzersizdir. KML tüm hematolojik malign hastalıklar içinde ilk defa spesifik kromozomal anomalisi gösterilmiş hastalıktır. KML’li hastaların %95’inden fazlasında, kromozom 9 ve 22’de yer değiştirme t(9;22) ile ortaya çıkan Philadelphia kromozomu taşıyan tek bir kök hücresinin klonal genişlemesi görülmektedir. Bu yer değiştirme, kromozom 9’daki abi genini (abelson lösemi virüsü) kromozom n’deki ber (breakpoint cluster region) genine bağlar ve sonuçta berablolarak adlandırılan bir onkogen meydana gelir. Bu onkogenin ürettiği ber-abl proteini yapısalolarak aktif bir tirozin kinazdır ve hematopoetik kök hücrelerinde lösemi gelişimine neden olmaktadır.
Bcr-abl füzyon proteini, sitokin düzenlemesinden ve kemik iliği stromasının etkisinden bağımsız olarak hücre büyümesine izin veren sinyal ileti mekanizmalarını aktif hale geçirir. Hücreler kemoterapiye ve normal programlanmış hücre ölümüne (apoptoz) dirençli hale gelirler. KML’li hastaların Philadelphia kromozomu tespit edilemeyen bir alt grubunda, reverse transkriptaz-polimeraz zincir reaksiyonu (RT-PCR) ile ber-abl yer değiştirme füzyon ürünlerinin saptanması, subkromozomal bir yer değiştirmenin de aynı patolojik genle sonuçlanabildiğini göstermektedir. KML tanısı karyotipleme, PCR veya floresan in situ hibritleme (FISH) ile Philadelphia kromozomunun tespit edilmesi ile konmaktadır.
Philadelphia kromozomunun tespiti, tanının daha kolay konulmasını ve hastalığın izlenebilmesini sağlamıştır. Çok duyarlı ve kantitatif olan RT-PCR tekniği hem 105- 106 periferik hücre arasından ber-abl-pozitif olan tek bir hücreyi dahi tespit edebilmekte hem de kan ve kemik iliği örneklerinden hastalığın derecesini belirlemeye olanak tanımaktadır. KML’de tedaviye cevap hematolojik (periferik kan sayımının normale dönmesi), sitogenetik (Philadelphia kromozomunın normal karyotipik analiz veya FTSH ile ortadan kalktığının gösterilmesi) ve moleküler (ber-abi geninin RT-PCR ile kaybolduğunun gösterilmesi) iyileşmeler olarak tanımlanmaktadır.
KML, MPD’lerin en sık karşılaşılan tipidir. Tüm lösemi lerin% 15-20’sini oluşturmaktadır ve 100.000 kişiden birinde görülmektedir. Ortalama başlangıç yaşı 53 olup, her yaştan kişi etkilenebilmektedir. Hastaların %40’1 başlangıçta asemptomatiktir. Diğerlerinde; yorgunluk, letarji, nefes darlığı, kilo kaybı, çabuk morarma ve erken tokluk gözlenebilmektedir. Fizik muayenede genellikle splenomegali saptanır. Laboratuar değerleri; lökosit sayısında çok fazla artma (ortalama 170x 109/1t), düşük lökosit alkalin fosfataz düzeyi, yüksek ürik asit ile laktat dehidrojenaz düzeyi ve trombositoz şeklinde olmaktadır.
Kronik faz KML’de periferik yayma incelenmesi ile olgunlaşmamış myeloblastlar (genellikle %5’in altında), myelositler, metamyelositler, bazofiller, eozinofiller, bantlar ve nötrofiller gibi granülositik gelişimin tüm evrelerindeki miyeloid hücreler görülebilir. Tersine iokomoid reaksiyon olarak tanımlanan, akut enfeksiyon veya sepsis sonucu gelişen, reaktif granülositik hiperplastik durumlarında, yaymada olgun nötrofiller, bunun yanı sıra bantlar, az sayıda miyelosit, bazofil ve eozinofıl bulunur. KML de kemik iliğinde yoğun hiperselüler yapı ile gelişimin tüm evrelerindeki miyeloid hücrelerin yoğunluğu ve retükülin fibroz görülür. Philadelphia kronomozununun geleneksel veya moleküler testler ile saptanması da KML tanısını doğrulayacaktır.
KML’nin doğal seyri, akut blast krizlerine dönüşebilen kronik faz ile karakterizedir. Hastalar tipik olarak kronik fazda tanı alırlar, bu evre 3-5 yıl kadar sürebilmektedir. Periferik lökosit sayıları yükselmiştir (eozinofili ve bazofili <%20), az sayıda blast saptanabilir «%5). Periferik kan sayımına bakılarak hastaların bu süreçte semptomatik olduğu söylenebilir. Sonuçta, hastalık, ateş, kilo kaybı, splenomegalinin kötüleşmesi ve kemik iliği hücrelerinin hızlı döngüsüne bağlı kemik ağrısı ile karakterize hızlanmış faza girer. Tedaviye rağmen lökosit sayısı özellikle de blastlar (% 10-19 arasında) artar.
Periferik bazofil sayısının artması (%20) histamin üretimine yol açar; kaşıntı, diyare ve yüzde kızarma gibi semptomlar gözlenir. Hızlanmış fazda hastalarda splenomegalide artma, kalıcı trombositopeni veya trombositoz ve lökositoz ile kemik iliği hücrelerinde yeni klonal sitogenetik anomalilere rastlanabilir. KML’nin hlast krizi olarak adlandırılan son fazı, iliğin yerini %20 veya daha fazla blast hücrelerinin aldığı, ilikte ve periferde normalolgun hücre elemanlarının kaybolduğu, ekstramedüller blast oluşumunun gözlendiği akut lösemi durumuna geçişi işaret etmektedir. Birkaç hafta veya ay içinde hastalar ölmektedir.
Hastaların üçte ikisinde AML gelişirken, geri kalanında akut lenfoid lösemi gelişiyor olması, başlangıçtaki neoplastik hücrenin birçok hücre tipine farklılaşabilme yeteneği olan bir kök hücresi olduğunu kanıtlamaktadır

Tedavide eskiden hidroksiüre ve busulfan gibi oral kemoterapötik ajanlar, KML’nin kronik fazı sırasında hastalarda miyeloid hücre sayısını azaltmak için kullanılıyordu. Bu ilaçlar akut hastalık komplikasyonlarını azaltmakta ancak uzun dönem prognozu veya blast krizlerine dönüşümü etkilememekteydi. interferon-a kullanımıyla beraber, kronik evre KML hastalarının %60-80’inde hematolojik iyileşmeler elde edildi ve %20 30 hastada sitogenetik cevap yaratan ilk ilaç oldu. interferon-a ile sitogenetik iyileşme sağlanması yaşam beklentisini uzatmış; kemoterapi ile kombinasyonu cevabı daha da artırmıştır. İnterferon-a ile tedavi edilen hastaların çoğunda PCR ile her-ahI translokasyonu tespit edilmesine ve nüks riskinin devam etmesine rağmen, hematolojik ve sitogenetik iyileşme yıllarca devam etmektedir.
Saptanabilir her ahl pozitif hücrelerin varlığına rağmen interferon-a’nın hangi mekanizma ile hastalığı kontrol ettiği bilinmemektedir. Maalesef hızlanmış faza ve/veya blast krizine giren KML hastaları interferondan fayda görmezler ve bu hastalarda kullanılacak yüksek doz kemoterapi rejimIeri 6 aydan kısa süren geçici düzelmeler sağlayabilirler.
KML’nin tedavisinde kullanılan imatinib mesilat (Gleevec, önceleri STl-57l olarak bilinmekteydi)'ın geliştirilmesi kanser tedavisinde elde dilen ilk başarı olmuştur. Gleevec her-ahI trombosit kaynaklı büyüme faktörü (PDGF) ve e-kit reseptör tirozin kinazlar’ın kompetetif inhibitörüdür. Gleevec ile yapılan pre-klinik çalışmalar, her-ahI taşıyan KML hücre serilerinin ve progenitör hücrelerin in vitro büyümesinin durduğunu göstermiştir. Bu oral aktif tirozin kinaz inhibitörü ile ilk klinik denemeler 1998’de interferon-a’ya cevap vermeyen KML hastalarında yapılmıştır.
İlaç kontrol altında tutulabilen yan etkilerle iyi tolere edilmiş, 4 hafta boyunca günde 300 mg’dan yüksek dozlarla hematolojik iyileşme sağlanmış ve %33’ünde 8 hafta sonra sitogenetik iyileşme elde edilmiştir. Bu çarpıcı sonuçlar pek çok deneme ile doğrulanmıştır. KML’nin kronik fazında olan, yeni tanı konmuş ve tedavi başlanmamış hastalarda Gleevec’in, tedavi başarısı açısından interferon-a ve sitarabin’a üstün olduğu kabul edilmiştir. Gleevec hem si togeneti k ve hematolojik iyileşme sağlamakta hem de hastalığın hızlanmış faza ya da blast fazına dönüşümünü geciktirmektedir. Yüksek doz Gleevec ayrıca hızlanmış faza ya da blast fazına girmiş bazı hastalarda da geçici hematolojk ve sitogenetik iyileşmeler yapabilmektedir. Gleevec artık KML tedavisinde standart olarak kullanılmasına rağmen, ki yeni tanı almış kronik evre KML hastalarının %80’inden çoğunda tam sitogenetik iyileşme sağlar, hastalığı iyileştirmez. İnterferon tedavisine benzer bir biçimde, Gleevec ile tam sitogenetik iyileşme elde edilen hastaların. büyük çoğunluğunda her ahl pozitif lösemik KML kök hücrelerine rastlanır.
Dolayısıyla hastalığı kontrol etmek için Gleevec’i hayat boyu kullanmak gerekir. Üstelik Gleevec tedavisine, özellikle de ek sitogenetik anomalileri ve reseptör kinaz bölgesinde mutasyonu olan kronik evre KML’nin ileri evrelerinde bulunan hastalarda, direnç geliştiği çoktan rapor edilmiştir. Sonuç olarak Gleevec tedavisi ile mükemmel sonuç elde edilen KML hastalarında bile hastalığın ilerleme ve tedavinin başarısız olma riski vardır. Yüksek doz Gleevec ve Gleevec dirençli vakalarda kullanılabilecek daha etkili yeni her-ahI kinaz inhibitörlerinin kullanımı şu anda araştırma aşamasındadır.